文本挖掘在基因組注釋中的應用
2017-03-21 10:50:06,
中華醫學圖書情報雜志 2017年3期
,
基因組注釋是指利用生物信息學方法對基因組中所有基因的生物學功能進行高通量注釋,包括核苷酸級別的注釋、蛋白質級別的注釋以及流程級別的注釋[1]。目前,常規的基因組注釋方法存在步驟過于繁瑣、需要借助高精尖設備、人工操作存在誤差、 “同源-功能相似”只是一種假說、模體本身具有的層次性以及涉及的分析工具較多無法自動化操作等問題,得到的結果存在誤差[2]。隨著計算機技術的發展以及關于基因研究的生物醫學文獻數量的不斷增加,利用文本挖掘技術[3]對生物醫學文獻分析來實現對基因組注釋成為一種新的研究趨勢。
1 材料和方法
筆者利用WOS數據庫中的文獻作為研究的樣本來源,檢索策略為:TS=(gene annotation* OR genomic* annotation*) AND TS=(text mining OR literature mining),檢索時間為2016年10月19日,限定時間段在2000-2016年之間,得到328篇相關文獻。利用書目共現分析軟件BICOMB抽取相關文獻中的引文,選取出現頻次在15次及以上的引文,共得到16篇高被引論文(表1)。利用BICOMB構建高被引論文——來源文獻矩陣(該矩陣可反映高被引論文在來源文獻中的分布情況),然后將詞篇矩陣導入聚類分析軟件gCluto中進行高被引論文的同被引聚類分析。

表1 328篇來源文獻中的高被引論文(n=16,f>=15)
2 結果與分析
將同被引聚類分析結果用可視化圖像表示,山峰圖見圖1,棋盤圖見圖2。圖1中16篇高被引論文根據其在328篇來源文獻中的被引情況可分成3個大類;圖2中行聚類是對于高被引論文的聚類,列聚類是對于來源文獻的聚類。圖2中行聚類結果也表明該16篇高被引論文可分為3類,表示文本挖掘技術在基因組注釋中的3個應用方向。各大類對應的高被引論文見表2。其中每個大類的內容可根據該大類中包含的高被引論文及其間的樹狀關系進行總結,通過對每個大類對應的列聚類中描述度較高的來源文獻(即每個類的類標簽文獻)的閱讀研究進一步把握各大類的內容。本文結合同被引論文聚類分析結果和各類中高被引論文,將文本挖掘技術在基因組注釋方面的應用分為權威工具的使用、文本挖掘工具和算法的開發、文本挖掘工具的檢驗3類。
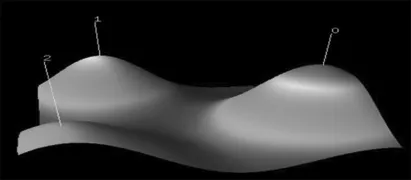
圖1 高被引論文聚類分析的山峰圖
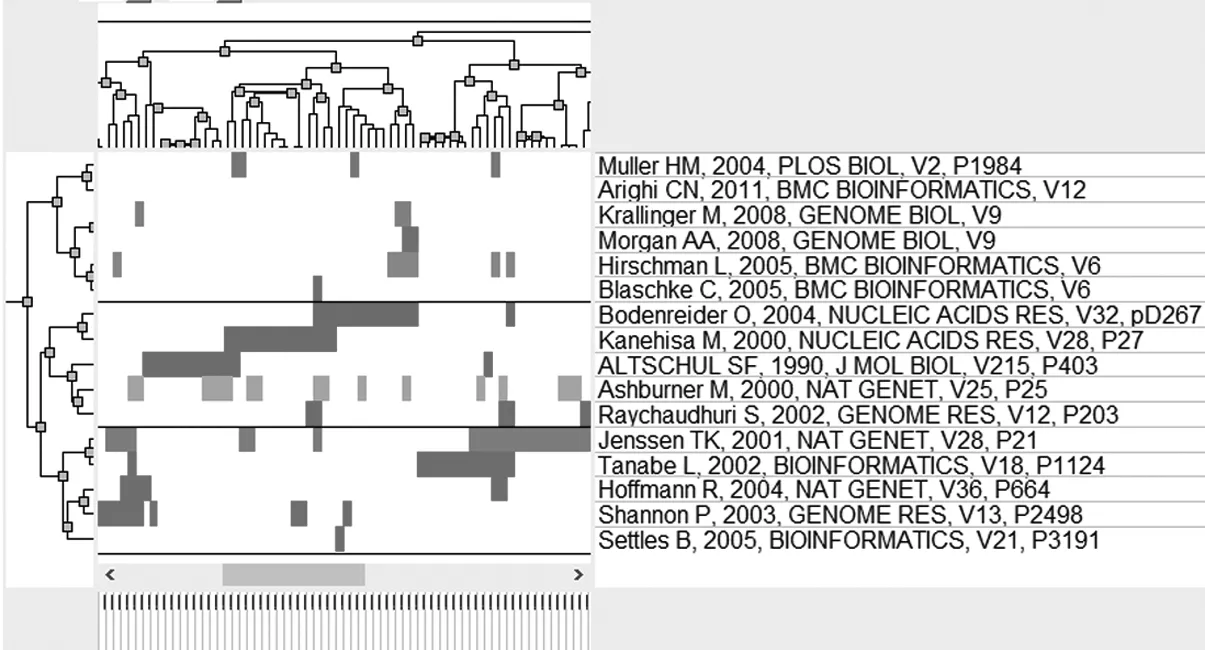
圖2 高被引論文聚類分析的棋盤圖

表2 3類對應的高被引論文
一是權威工具的使用。通過對Cluster 1中相關高被引文獻以及類標簽文獻的分析,總結出在基因組注釋的相關研究中,收錄有基因組及基因產物相關序列、結構或功能信息的數據庫和軟件工具以及與基因相關的受控詞匯表被廣泛利用,如京都基因和基因組百科全書(Kyoto Encyclopedia of Genomes, KEGG)[4-5]、一體化醫學語言系統(The Unified Medical Language System, UMLS)[6-7]、基因本體(Gene Ontology, GO)[8]、基本局域聯配搜索工具(Basic Local Alignment Tool, BLAST)等。這些數據庫、軟件將已知的基因相關信息匯總、整理并組織起來,提供給科研人員使用和查詢。Taniya T等人[9]在尋找特定復雜疾病新的候選基因的研究中利用了京都基因和基因組百科全書、基因本體以及其他一些數據庫中的信息來獲取與類風濕性關節炎和前列腺癌相關的已知致病基因。
然而這些數據庫或軟件工具中有些關于基因、蛋白質等物質的注釋信息基本依賴于專家人工從文獻集中獲得。隨著生物醫學科技文獻數量的增加以及用戶需求的增加,這種數據收集方法缺乏靈活性,其收錄信息的范圍也受到限制。因此從文獻中自動提取信息的計算機算法被開發出來作為人工開發數據庫的補充,尤其是基因概念之間的關聯研究及應用[10-11]。
二是文本挖掘工具和算法的開發。對Cluster 2中相關高被引文獻進行分析,五篇高被引論文的研究方向都是對于文本挖掘工具的介紹,包括基因和蛋白質等相關實體的識別工具[12-13]、基因共現網絡創建工具[14]、利用基因與蛋白作為鏈接點構建文獻網絡的信息系統[15]等等。在此基礎上再對Cluster 2中的類標簽文獻進行分析,我們總結出在基因組注釋中,相關文本挖掘工具和算法的開發與利用是文本挖掘技術在基因組注釋方面的一大重要應用。
在分子生物學及相關領域,大規模高通量實驗技術的發展和生物信息學工具的使用產生了大量的數據并促進了科學文獻的增長,但也使得許多顯性或隱性知識被掩蓋在文獻中難以被科研人員利用,這促進了文本挖掘工具和算法的發展與利用[16]。通過Rodriguez-Esteban R等人[17]與Krallinger M等人[18]對于生物醫學領域文本挖掘技術的論述,我們可以總結出文本挖掘技術涉及到命名實體識別、關系檢測、知識發現等多個階段,在各個階段中都有相關的文本挖掘工具或應用程序被開發出來。比如在命名實體識別階段,有Whatizit系統(一個文本處理系統,可以識別文本中的分子生物學術語,并將其鏈接到公共可用的數據庫中)、ABNER程序(A Biomedical Named Entity Recognizer,生物醫學命名實體識別器,是一個可以識別蛋白質、DNA、RNA、細胞系和細胞類型這五種術語的開源軟件工具)等工具;在關系檢測階段,有MedGene(一種全面估計和總結Medline中所有人類基因——疾病關系相對強度的文本挖掘工具)等工具,并且基因本體和蛋白質相互作用網絡也能分別展示相關基因、蛋白質的親疏遠近關系;在知識發現階段,有Arrowsmith(一個免費的、基于公共網絡的兩節點搜索工具,允許用戶在PubMed中識別任何兩組文章集之間有生物學意義的連接)等工具。
三是文本挖掘工具的檢驗。對Cluster 0中相關高被引文獻進行分析,6篇高被引論文中有5篇文獻的主要內容是對于BioCreative(Critical Assessment of Information Extraction systems in Biology,生物學中信息提取系統的嚴格評價)評估的描述[19-23],再結合對Cluster 0中描述度較高的類標簽文獻的分析,發現文本挖掘在基因組注釋中的一大應用是進行文本挖掘競賽以檢驗各文本挖掘工具。
在生物醫學領域,已有很多關于基因、蛋白等物質的注釋數據庫被開發。隨著生物醫學領域科技文獻量的增長,依靠專家人工從文獻中提取有用信息策展相關數據庫在時間上已經有很大的局限性,這促進了生物醫學領域文本挖掘技術尤其是自然語言處理技術的發展,也使得BioCreative評估應運而生。 BioCreative評估建立于2004年,主要目的在于評估應用于生物醫學領域的文本挖掘技術的最高水平。除此之外,該評估還促進了相關數據庫開發者與文本挖掘研究人員之間的交流,有利于自動化的文本挖掘技術與人工策展相結合共同進行數據庫的開發。 從2004年開始,BioCreative評估用來檢驗各文本挖掘工具的任務多圍繞文獻中基因、蛋白質等相關實體的提取、基因標準化、利用基因本體或蛋白質相互作用網絡在全文中提取基因或蛋白質的功能注釋等方面展開,在這期間還邀請文本挖掘工具最終用戶參與進來,加強文本挖掘工具解決生物醫學研究中實際問題的能力[18,24-26]。
3 討論
本文通過對WOS中有關文本挖掘與基因組注釋的相關文獻的檢索、篩選、聚類和閱讀研究,發現文本挖掘技術在基因組注釋方面的應用大致分為權威工具的使用、文本挖掘工具和算法的開發、文本挖掘工具的檢驗3方面。伴隨著生物醫學文獻量的不斷增加、高通量實驗技術的不斷進步以及科研人員對于信息提取工具需求的增加,相信會有越來越多的文本挖掘工具被開發出來。與此同時,隨著文本挖掘工具競賽的舉辦,其研發會越來越貼近科研人員的現實需要。對于依靠人工從文本集中收集有用信息的數據庫等工具的研發,未來的發展趨勢應該會將文本挖掘技術整合進相關開發流程,更加依賴文本挖掘技術來提取信息以充實數據庫。當然,除了在基因組注釋方面,文本挖掘技術在藥物重定位研究、藥物靶向位點研究等其他生物醫學領域也會發揮越來越重要的作用。
猜你喜歡
甘肅教育(2020年8期)2020-06-11 06:10:02
制造技術與機床(2019年10期)2019-10-26 02:48:08
電子制作(2018年18期)2018-11-14 01:48:06
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
小學教學參考(2015年20期)2016-01-15 08:44:38
人間(2015年20期)2016-01-04 12:47:10
