血色病合并貧血、重度黃疸1例報告
2017-04-24 03:37:10李東穎杜白雪李建生
臨床肝膽病雜志 2017年4期
朱 敏, 李東穎, 杜白雪, 李建生
(鄭州大學第一附屬醫院 消化內科, 鄭州 450052)
血色病合并貧血、重度黃疸1例報告
朱 敏, 李東穎, 杜白雪, 李建生
(鄭州大學第一附屬醫院 消化內科, 鄭州 450052)
血色素沉著癥; 貧血; 黃疸; 病例報告
遺傳性血色病(hereditary hemochromatosis,HH)是由鐵代謝紊亂引起體內鐵負荷過多所致的一種常染色體院性遺傳疾病。臨床表現為乏力、倦怠、糖尿病、皮膚色素沉著癥、肝硬化、心肌病、關節炎等。HH的地理分布呈世界性,在北歐日耳曼和高加索人群中的發病率約1/220~1/250[1-2],在亞太地區較少見。
1 病例資料
患者男性,63歲,因“反復黃疸40余年,加重1 d”入本院。查體:全身皮膚、鞏膜黃染,未見明顯皮膚色素沉著,淺表淋巴結未觸及,瞼結膜無明顯蒼白,腹直肌外緣可見一長約15 cm手術后疤痕,肝臟肋緣下未觸及,脾臟肋緣下可觸及,平臍,右側未過腹正中線,余查體未見異常。既往史及個人史:幼時即發現脾大,約9歲發現貧血,給予口服藥物治療(具體不詳),此后至2015年期間是否存在貧血不詳。1年前曾患膽囊結石、膽總管結石,行膽囊切除術+膽總管切開取石T管引流術,術后再次出現膽紅素升高。近1年間斷復查肝功能,膽紅素均偏高(具體不詳),因貧血較重,給予輸注懸浮紅細胞治療4次(400 ml/次,共1600 ml)。否認長期大量飲酒史,但近期有服用肝損傷藥物史(入院前約1周曾因牙痛口服布洛芬緩釋片,0.3 g、2次/d,共服3 d)。家族史:父親因慢性乙型肝炎肝硬化并發癥去世(具體不詳),母親因心臟病猝死(具體不詳),3姐1弟1妹、1子體健,否認家族遺傳疾病史。
實驗室檢查:血常規:Hb 106 g/L,WBC 6.7×109/L,RBC 3.37×1012/L,PLT 139×109/L,紅細胞壓積 0.296,平均血紅蛋白濃度 357 g/L,網織紅細胞比例12.99%,網織紅細胞計數0.361×1012/L;尿常規:膽紅素陽性(+++);空腹血糖 7.25 mmol/L(飲食控制后復測空腹血糖正常,后患者拒絕進一步行口服葡萄糖耐量試驗檢查);肝功能:ALT 81 U/L,AST 46 U/L,GGT 398 U/L,TBil 597.2 μmol/L,DBil 464.6 μmol/L,IBil 132.6 μmol/L,TBA 214.1 μmol/L;CA12-5 17.87 μ/ml,CA19-9 146.9 μ/ml;鐵三項:鐵蛋白 1708.3 ng/ml,血清鐵 31.1 μmol/L,總鐵結合力率 31.32 μmol/L,不飽和鐵結合力 0.22 μmol/L。肝炎病毒抗體(HAV、HBV、HCV、 HEV)均陰性;特殊病毒抗體(人類皰疹病毒、巨細胞病毒、單純皰疹病毒等)均陰性;自身免疫性肝病相關抗體均陰性;銅藍蛋白、凝血功能未見異常。肝膽胰脾彩超:肝臟彌漫性回聲改變,脾大。腹部16層CT平掃增強(圖1,2):(1)脾大、脾梗死、脾內鈣化灶;(2)膽囊術后改變。磁共振胰膽管造影(圖3,4):(1)脾低信號、符合血色病;(2)脾大;(3)膽囊缺如﹑術后表現;(4)肝內外膽管未見明顯異常。 結合鐵蛋白升高及肝臟CT、MRI表現,初步診斷為血色病,為進一步明確診斷及篩查其他組織器官受累情況,完善肝活組織病理檢查、基因檢查等,肝臟病理(圖5,6):肝小葉結構基本存在,肝細胞輕度灶性大泡性脂肪變性,肝細胞內見大量含鐵血黃素沉積及部分膽色素沉著,匯管區淋巴細胞等炎細胞浸潤,纖維組織輕度增生,G1/S1。免疫組化:HBsAg陰性。特殊染色:Masson染色陽性、網染陽性、鐵染色陽性。骨髓穿刺:骨髓增生Ⅲ度(活躍),粒系成熟階段比值略低于正常,形態未見異常,幼稚階段比值形態大致正常。紅系增生明顯活躍,晚幼紅細胞比值明顯高于正常,并可見花瓣核,成熟紅細胞大小不等,以小細胞為主。全片巨核細胞9個,7個為成熟顆粒型、1個為成熟產板型、1個為裸核型,片中血小冊板較少。血片有核細胞分類大致正常,成熟紅細胞大小不等,以小細胞為主。基因檢測(PCR結合直接測序法):HFE基因及SLC40A1基因外顯子編碼區域未檢測到致病突變。外院電子胃鏡未見食管胃底靜脈曲張。甲狀腺功能、心臟彩超未見異常。
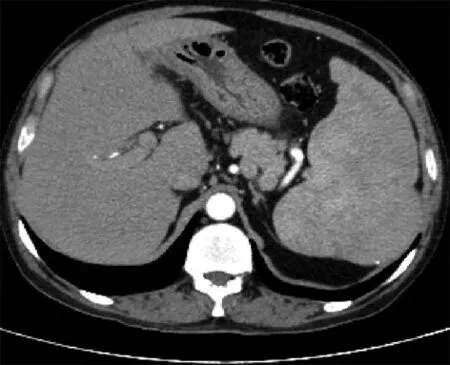
圖2 CT增強 肝臟密度普遍增高,120 KV時最大CT值155 HU,脾大

圖3 磁共振胰膽管成像T1加權像 肝脾普遍信號減低,脾大
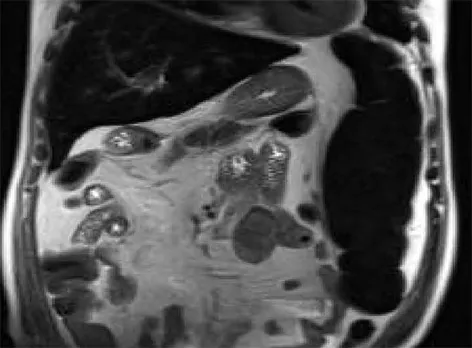
圖4 磁共振胰膽管成像冠位T2加權像 肝脾普遍信號減低,脾大
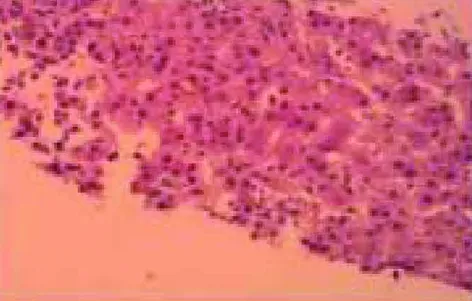
圖5 HE染色示肝細胞內色素顆粒沉著(×400)

圖6 鐵染色示肝細胞內鐵沉積(×400)
2 討論
HH是常染色體遺傳病,根據基因突變類型不同,又可以分為HFE相關HH和非HFE相關HH。HFE相關HH即Ⅰ型HH, 其中最常見的兩種錯義突變類型為C282Y和H63D。據美國肝病學會報道,約80%~85%的HH患者為C282Y純合突變,而H63D僅在與C282Y形成雜合突變時才可能導致血色病[1]。已報道的非HFE相關HH的基因有HJV(ⅡA型)、HAMP(ⅡB型)、TFR2(Ⅲ型)、SLC40A1(Ⅳ型)[1]。
本例患者以黃疸為主訴入院,但因缺乏進一步的檢查,發展為血色病的原因尚不能完全肯定。行基因檢測,可排除Ⅰ型及Ⅳ型HH。雖然Ⅱ、Ⅲ型HH非常罕見,但呂婷霞等[3]研究指出,我國HH患者的突變類型可能以非Ⅰ型HH為主,與歐美國家差別較大,并且可能存在與目前已知基因不同的HH相關基因參與了鐵代謝的調節過程,因此不能完全排除HH可能。此外,繼發性血色病發病原因復雜,結合本例患者特點,慢性貧血疾病、腸外鐵過載[4]導致的繼發性血色病也不能完全排除。本例重度黃疸的原因考慮為多因素共同作用的結果,即血色病基礎上服用肝損傷藥物所致。貧血的原因方面,結合患者幼時就發現脾大、貧血,反復黃疸數十年,入院后骨穿刺檢查結果,考慮與溶血相關,但因多種因素未能進一步行抗人球蛋白試驗、糖水試驗、尿含鐵血黃素試驗、外周血細胞形態分析等檢查明確,甚憾。本例患者確診為血色病后因合并貧血,不宜行靜脈放血治療,故建議鐵螯合劑治療,但因經濟原因拒絕,遂給予保肝、降酶、退黃等內科綜合治療后癥狀好轉出院。
血色病在亞太地區發病率低,起病隱匿,臨床癥狀不典型,容易誤診或漏診,需引起內科醫師的重視,早期診斷并給予及時正確的治療可提高患者的生存率和生活質量。
[1] BACON BR, ADAMS PC, KOWDLEY KV, et al. Diagnosis and management of hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases[J]. Hepatology, 2011, 54(1): 328-343.
[2] WOOD MJ, SKOIEN R, POWELL LW. The global burden of iron overload[J]. Hepatol Int, 2009, 3(3): 434-444.
[3] LYU TX, ZHANG W, LI XJ, et al.Characteristics of gene mutation in Chinese patients with hereditary hemochromatosis[J]. J Clin Hepatol, 2016, 32(8): 1571-1574. (in Chinese) 呂婷霞, 張偉, 李瀟瑾, 等. 我國人群遺傳性血色病基因突變特點分析[J]. 臨床肝膽病雜志, 2016, 32(8): 1571-1574.
[4] BOTTOMLEY SS. Secondary iron overload disorders[J]. Semin Hematol, 1998, 35(1): 77-86.
引證本文:ZHU M, LI DY, DU BX, et al. A case of hemochromatosis with anemia and severe jaundice[J]. J Clin Hepatol, 2017, 33(4): 742-743. (in Chinese) 朱敏, 李東穎, 杜白雪, 等. 血色病合并貧血、重度黃疸1例報告[J]. 臨床肝膽病雜志, 2017, 33(4): 742-743.
(本文編輯:劉曉紅)
A case of hemochromatosis with anemia and severe jaundice
ZHUMin,LIDongying,DUBaixue,etal.
(DepartmentofGastroenterology,TheFirstAffiliatedHospitalofZhengzhouUniversity,Zhengzhou450052,China)
hemochromatosis; anemia; jaundice; case reports
10.3969/j.issn.1001-5256.2017.04.033
2016-08-18;
2016-12-09。
朱敏(1989-),女,主要從事消化系統疾病的診治與研究。
李建生,電子信箱:lijiansheng@medmail.com.cn。
R589
B
1001-5256(2017)04-0742-02
