ABT-263中間體羧酸片段合成研究
2017-04-27 01:36:51申煥玲周國春
四川輕化工大學學報(自然科學版) 2017年2期
申煥玲, 周國春
(南京工業大學藥學院, 南京210009)
ABT-263中間體羧酸片段合成研究
申煥玲, 周國春
(南京工業大學藥學院, 南京210009)
報道了Bcl-2家族蛋白抑制劑ABT-263(1)中間體羧酸片段即4-(4-((2-(4-氯苯基)-5,5-二甲基環己基-1-烯)甲基)哌嗪-1-基)苯甲酸(2)的合成方法。首先4,4-二甲基環己酮與三溴化磷和N,N-二甲基甲酰胺反應生成第一個中間體化合物2-溴-5,5-二甲基環己基-1-烯甲醛(3),經Suzuki偶聯反應,還原、鹵代、胺化,最后經水解、酸化得到目標產物(2)。經過6步反應,使用廉價易得的原料和試劑,反應條件溫和,后處理方便,純化方法簡單,總產率達到48.0%,明顯高于現有文獻報道產率,適合大規模生產。各步反應產物結構經過1H NMR確認。
ABT-263;羧酸片段;合成
引言
細胞凋亡或程序性細胞死亡是通過消除損壞和老化的細胞而保持機體正常的發育和組織的完整性[1-3]。在這個調節過程中的任何改變都將引發包括癌癥在內的一系列疾病。Bcl-2家族蛋白是細胞凋亡的關鍵調節因子,在調節細胞凋亡信號中發揮重要作用[4-6]。因此,將Bcl-2家族蛋白作為潛在的治療藥物靶點成為現今抗癌藥物研究的熱點,進而研發出一類前景較好的抗癌化合物[7-8](圖1)。Wang[9-15]課題組報道了一系列化合物,如棉子酚[9]、TW-37[10]、TM-1206[12]等;René Grée[16-18]課題組也報道了一些化合物,如三唑衍生物[16]等;雅培制藥(Abbott Laboratories)[19-21]報道了ABT-737[19]、ABT-263[20-21](1)等活性化合物。雖然這些化合物都存在口服生物利用度低,給藥困難等問題,但研究表明ABT-263口服生物利用度高于同類型的其他活性化合物,因此受到了廣泛的關注。
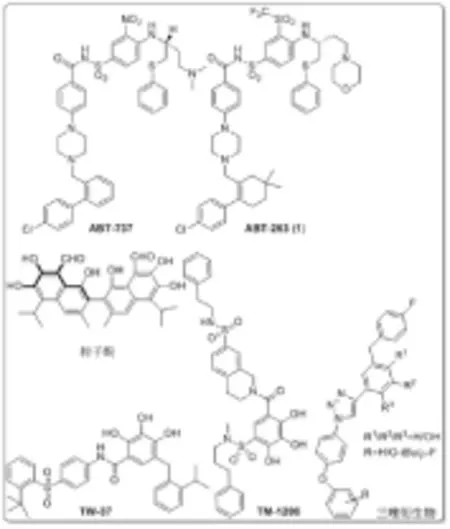
圖1以Bcl-2家族蛋白為藥物靶點的重要分子
ABT-263的逆合成分析[20](圖2)顯示化合物(1)由羧酸部分化合物(2)和苯磺酰胺部分兩個中間體縮合而成,本文的研究目的是改進ABT-263羧酸部分化合物(2)的合成方法。自化合物(2)被報道以來,近幾年合成方法主要有雅培和藥明康德兩條合成路線。在雅培制藥公司合成化合物(2)的過程中[20],還原胺化產率只有38.0%,并且使用了毒性較大的氰基硼氫化鈉,總收率只達到了14.0%,而且大多中間產物分離純化困難。藥明康德公司的合成路線[22],由于溶劑殘留特別是水份對下一步鹵代反應會有較明顯的不良影響,而醛和醇的沸點都較低(實驗研究發現在油泵真空干燥時醛和醇在5小時內分別減重85.4%和70.2%左右),因此在大規模生產過程中產品的干燥可能比較困難;另外,該工藝中胺烷基化和偶聯反應的總收率只有62.0%,而且偶聯反應需要三種混合溶劑,雖然使用貴金屬催化劑的偶聯反應放在后面合成階段,成本上較為合理,但胺的偶聯中用到了大量的較貴的碘化鈉(3.04eq),這樣也使成本上升。與雅培制藥公司工藝相比雖然產率提高了,但是仍然有較多不足之處。
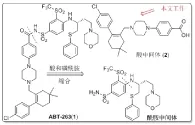
圖2ABT-263逆合成分析
綜合以上兩條合成路線[20-22],本文提出以4,4-二甲基環己酮為原料,與三溴化磷和N,N-二甲基甲酰胺反應合成化合物(3),經過簡單處理的化合物(3)(前一步的溶劑殘留對下一步反應幾乎沒有影響)可以直接進行Suzuki偶聯反應,然后進行醛還原、烯丙位伯醇氯代、胺烷基化,最后酯水解和酸化得到終產物(2)(圖3)。
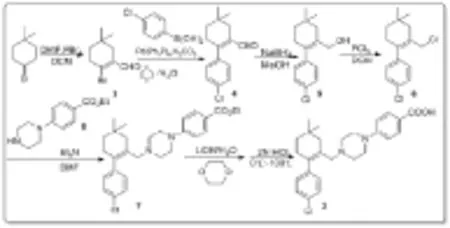
圖3化合物(2)的合成路線
1實驗部分
1.1試劑與儀器
DZF-6050真空干燥器(上海一恒科學儀器有限公司);旋轉蒸發器RE-5299,DF-101S型集熱式恒溫加熱磁力攪拌器(鞏義市予華儀器有限責任公司);Bruker-400 MHz型核磁共振儀(瑞士Bruker公司);Agilent LS/MS型高分辨質譜議(HRMS)(美國Agilent公司)。
4-(哌嗪-1-基)苯甲酸乙酯(8)按照文獻[23]方法合成;4,4-二甲基環己酮(試劑級,安微濟達化工有限公司);四三苯基磷鈀(試劑級,薩恩化學技術(上海)有限公司);三溴化磷、哌嗪、4-氟苯甲酸乙酯、4-氯苯硼酸(試劑級,基麗化學技術(上海)有限公司);N,N-二甲基甲酰胺、二氯甲烷、乙酸乙酯、甲醇、1,4-二氧六環、石油醚(分析純,西隴化工股份有限公司);其他無機試劑均為分析純,上海凌峰化學試劑有限公司。
1.2合成
1.2.14-(哌嗪-1-基)苯甲酸乙酯(8)的合成
稱取哌嗪12.29 g(142.7 mmol)和K2CO313.15 g(95.14 mmol)置于250 mL的三口燒瓶中,N2保護,加入150 mL二甲基亞砜。預冷,緩慢加入4-氟苯甲酸乙酯8.0 g(47.57 mmol)。滴加完畢后,升溫至120 ℃。攪拌6 h,TLC監測反應無原料,結束反應。冷卻至室溫,將體系緩慢加入到冷水中,乙酸乙酯萃取多次,依次用蒸餾水、飽和NaHCO3水溶液、飽和NaCl水溶液洗滌兩次,有機相用無水Na2SO4干燥,過濾,濾液濃縮后得化合物(8)[23]10.48 g,產率為94.1%。1H NMR(400 MHz,(CD3)2SO):δ7.77(d,J=9.1 Hz,2H,4,6-H),6.95(d,J=9.1 Hz,2H,1,3-H),4.23(q,J=7.1 Hz,2H,17-H),3.19~3.22(m,4H,8,12-H),2.79~2.81(m,4H,9,11-H),1.28(t,J=7.1 Hz,3H,16-H)。無需純化,直接投下一步。
1.2.22-溴-5,5-二甲基環己基-1-烯甲醛(3)的合成
N2保護,0 ℃下,將20.20 mL三溴化磷(216.34 mmol)緩慢滴加到150 mL無水二氯甲烷和18.80 mL無水N,N-二甲基甲酰胺(246.18 mmol)的混合溶液中,滴加完畢后,體系慢慢恢復至室溫,并在此溫度下攪拌0.5 h。再次冷卻至0 ℃,將4,4-二甲基環己酮9.40 g(74.60 mmol)的80 mL無水無氧二氯甲烷溶液緩慢滴入到上述體系中,滴加完畢后,慢慢恢復至室溫,此時加上尾氣吸收處理裝置,并攪拌過夜。TLC監測反應無原料,結束反應。將反應液緩慢倒入到冰冷的飽和NaHCO3水溶液中,石油醚萃取,后處理方式與1.2.1相同。濃縮后得黃色油狀液體化合物(3)[20-22]10.47 g,產率65.0%。1H NMR(400 MHz,CDCl3):δ10.01(s,1H,10-H),2.73~2.76(m,2H,2-H),2.07(s,2H,5-H),1.51(t,J=6.5 Hz,2H,1-H),0.93(s,6H,7,8-H)。無需純化,直接投下一步。
1.2.32-對氯苯基-5,5-二甲基環己基-1-烯甲醛(4)的合成
N2保護,0 ℃下, 將干燥后的3 10.47 g(48.47 mmol)的145 mL 1,4-二氧六環溶液緩慢加入到4-氯苯硼酸7.58 g(48.47 mmol)與四三苯基磷鈀151 mg (0.13 mmol)的混合物中,滴加完畢,升溫至100 ℃,小心分批加入145 mL K2CO313.38 g(96.94 mmol)水溶液,繼續攪拌2 h,TLC監測反應無原料,結束反應。體系冷卻至室溫,乙酸乙酯萃取,后處理方式與1.2.1相同。過濾,濾液濃縮后用硅膠柱層析純化(石油醚/乙酸乙酯=15/1)得黃色油狀液體化合物(4)[24]10.34 g,產率86.0%。1H NMR (400 MHz,CDCl3):δ9.49(s,1H,16-H),7.36(d,J=8.5 Hz,2H,12,14-H),7.16(d,J=8.5 Hz,2H,11,15-H),2.52~2.56(m,2H,2-H),2.14(s,2H,5-H),1.53(t,J=6.4 Hz,2H,1-H),0.99(s,6H,7,8-H)。
1.2.4(2-對氯苯基-5,5-二甲基環己基-1-烯)甲醇(5)的合成
0 ℃下,將化合物(4)10.34 g(41.69 mmol)溶于125 mL甲醇中,加入硼氫化鈉1.90 g(50.03 mmol)。保持0 ℃,攪拌2 h。TLC監測反應無原料,結束反應,將體系緩慢倒入冰水中,攪拌0.5 h,乙酸乙酯萃取,后處理方式與1.2.1相同。濃縮后得淡黃色油狀液體化合物(5)[22,25-26]10.42 g,產率100%。1H NMR(400 MHz,(CD3)2SO):δ7.36(d,J=8.4 Hz,2H,12,14-H),7.21(d,J=8.4 Hz,2H,11,15-H),4.52(t,J=5.3 Hz,1H,17-H),3.68(d,J=5.2 Hz,2H,10-H),2.23(t,J=5.9 Hz,2H,2-H),1.98(s,2H,5-H),1.41(t,J=6.4 Hz,2H,1-H),0.95(s,6H,7,8-H)。無需純化,直接投下一步。
1.2.5(2-對氯苯基-5,5-二甲基環己基-1-烯)氯甲烷(6)的合成
0 ℃下,將化合物(5) 10.42 g(41.68 mmol)溶于125 mL無水二氯甲烷,然后快速稱量并加入五氯化磷4.33 g(20.84 mmol)。攪拌4 h,TLC監測反應無原料,結束反應。將體系緩慢倒入冰水中,攪拌至澄清透明,乙酸乙酯萃取,后處理方式與1.2.1相同。濃縮后得無色液體化合物(6)[22,26]10.52 g,產率為94.2%。1H NMR (400 MHz,CDCl3):δ 7.32 (d,J=8.39 Hz,2H,12,14-H),7.16(d,J=8.39 Hz,2H,11,15-H),3.88(s,2H,10-H),2.30(t,J=6.38 Hz,2H,2-H),2.05(s,2H,5-H),1.48(t,J=6.46 Hz,2H,1-H),1.00(s,6H,7,8-H)。無需純化,直接投下一步。
1.2.64-(4-((2-(4-氯苯基)-5,5-二甲基環己基-1-烯)甲基)哌嗪-1-基)苯甲酸乙酯(7)的合成
常溫下,將化合物(6)10.52 g(39.25 mmol)、化合物(8)9.185 g(39.25 mmol)溶于118 mLN,N-二甲基甲酰胺,緩慢加入三乙胺13.68 mL(98.12 mmol),升溫至80 ℃。攪拌3 h,TLC監測反應無原料,結束反應。將體系緩慢倒入冰水中,攪拌至澄清透明,乙酸乙酯萃取,后處理方式與1.2.1相同。濃縮后得白色固體化合物(7)[22,26]17.56 g,產率96.0%。1H NMR(400 MHz,CDCl3):δ7.89(d,J=9.0 Hz,2H,25,27-H),7.28(d,J=1.7 Hz,2H,12,14-H),7.00(d,J=8.4 Hz,2H,11,15-H),6.81(d,J=9.0 Hz,2H,24,28-H),4.32(q,J=7.1 Hz,2H,33-H),3.25~3.27(m,4H, 19,21-H),2.80(s,2H,10-H),2.34~2.37(m,4H,18,22-H),2.26(t,J=5.3 Hz,2H,2-H),2.02(s,2H,5-H),1.47(t,J=6.5 Hz,2H,1-H),1.36(t,J=7.1 Hz,3H,32-H),0.99(s,6H,7,8-H)。無需純化,直接投下一步。
1.2.74-(4-((2-(4-氯苯基)-5,5-二甲基環己基-1-烯)甲基)哌嗪-1-基)苯甲酸(2)的合成
將化合物(7)17.56 g(37.68 mmol)溶于435 mL 1,4-二氧六環。預冷,緩慢將87 mL 1N氫氧化鋰水溶液滴加至上述體系,升溫至100 ℃。攪拌24 h,TLC監測反應無原料,結束反應。體系冷卻至室溫,減壓蒸餾除去1,4-二氧六環,析出大量白色固體,重新溶于水,1N鹽酸調節pH≈6。抽濾得濾餅,并用蒸餾水洗滌,于30 ℃真空干燥8 h,得白色固體純品化合物(2)[20]15.70 g,產率95.1%。1H NMR(400 MHz,(CD3)2SO):δ12.25(s,1H,31-H),7.73(d,J=8.7 Hz,2H,25,27-H),7.37(d,J=8.2 Hz,2H,12,14-H),7.12(d,J=8.2 Hz,2H,11,15-H),6.90(d,J=8.8 Hz,2H,24,28-H),3.21~3.24(m,4H,19,21-H),2.74(s,2H,10-H),2.25~2.28(m,4H,18,22-H),2.20~2.24(m,2H,2-H),1.99(s,2H,5-H),1.43(t,J=6.1 Hz,2H,1-H),0.97(s,6H,7,8-H)。HR-ESI-MSm/z:439.2147 C26H32N2O2Cl{[M+H]+}。
2結果與討論
與雅培制藥工藝相比[20-21],無需純化,就可將還原胺化由原來的38.0%提高到90.4%,從而提高了總產率。
與藥明康德相比[22],(1)將Suzuki偶聯反應放到第二步,不僅克服醛和醇的低沸點且難干燥問題,而且使Suzuki偶聯活性增加,重金屬催化劑從原來的5.0%當量[24]降低到了2.7‰當量,并且使用常規反應溶劑;(2)從4.6當量三溴化磷[22]改成0.5當量的五氯化磷,總磷量降低了;(3)在鹵代反應中用氯代代替了原來的溴代,由于處于烯丙位的氯代物穩定性大于溴代物,降低了烯丙位鹵代物的分解副反應使得氯代物收率提高(氯代物94.2%,溴代物86.0%);(4)氯代物與胺反應時不需要NaI或其他活化劑的存在下,就可以直接發生胺烷基化反應;同樣由于氯代物的穩定性降低了副反應,胺烷基化產率由溴代物的70.0%[22]提高到了氯代物的96.0%。
3結束語
綜上所述,本文合成路線反應條件簡單,反應溶劑常規易得,僅需一步純化,總產率就可達到48.0%,具有操作簡單、產率高、反應條件溫和等優點。
[1] Lee E F,Clarke O B,Evangelista M,et al.Discovery and molecular characterization of a Bcl-2-regulated cell death pathway in schistosomes[J].Proc Natl Acad Sci USA,2011,108(17):6999-7003.
[2] 李功權.Mcl:1和IAPs家族蛋白介導肝癌細胞抗凋亡作用及其分子機制的研究[D].鄭州:鄭州第一臨床學院,2014.
[3] ADAMS J M,CORY S.The Bcl-2 protein family:Arbiters of cell survival[J].Science,1998,281(5381):1322-1326.
[4] ADAMS J M,CORY S.The Bcl-2 apoptotic switch in cancer development and therapy[J].Oncogene,2007,26(9):1324-1337.
[5] GROSS A,MCDONNELL J M,KORSMEYER S J.BCL-2 family members and the mitochondria in apoptosis[J].Genes & Dev,1999,13(15):1899-1911.
[6] MARTINOU J C,YOULE R J.Mitochondria in Apoptosis:Bcl-2 Family Members and Mitochondrial Dynamics[J].Dev Cell,2011,21(1):92-101.
[7] VANDELFT M F,WEI A H,MASON K D,et al.The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized[J].Cancer Cell,2006,10(5):389-399.
[8] LETAI A.BCL-2:found bound and drugged[J].Trends Mol Med,2005,11(10):442-444.
[9] WANG S,MI(US) S,YANG D,et al.Small molecule antagonists of bcl-2 family proteins[P].US,20030008924,2003-06-09.
[10] WANG G P,NIKOLOVSKA-COLESKA Z,YANG C Y,et al.Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins[J].J Med Chem,2006,49(21):6139-6142.
[11] TANG G Z,DING K,NIKOLOVSKA-COLESKA Z,et al.Structure-based design of flavonoid compounds as a new class of small-molecule inhibitors of the anti-apoptotic bcl-2 proteins[J].J Med Chem,2007,50(14):3163-3166.
[12] TANG G Z,YANG C Y,NIKOLOVSKA-COLESKA Z,et al.Pyrogallol-based molecules as potent inhibitors of the antiapoptotic Bcl-2 proteins[J].J Med Chem,2007,50(8):1723-1726.
[13] TANG G Z,NIKOLOVSKA-COLESKA Z,QIU S,et al.Acylpyrogallols as inhibitors of antiapoptotic Bcl-2 proteins[J].J Med Chem,2008,51(4):717-720.
[14] ZHOU H B,CHEN J F,MEAGHER J L,et al.Design of Bcl-2 and Bcl-xL Inhibitors with Subnanomolar Binding Affinities Based upon a New Scaffold[J].J Med Chem,2012,55(10):4664-4682.
[15] BAI L C,CHEN J F,MCEACHERN D,et al.BM-1197:A Novel and Specific Bcl-2/Bcl-xL Inhibitor Inducing Complete and Long-Lasting Tumor Regression In Vivo[J].Plos One,2014,9(6):1-13.
[16] VO D D,GAUTIER F,BARILLE-NION S,etal.Design,synthesis and biological evaluation of new inhibitors of Bax/Bcl-xL interaction in cancer cells[J].Bioorg Med Chem Lett,2014,24(7):1758-1761.
[17] VO D D,GAUTIER F,BARILLE-NION S,et al.Synthesis of new mixed phenol/heterocyclic derivatives and studies of their activity as inhibitors of Bax/Bcl-xL interaction[J].Tetrahedron,2014,70(2):301-311.
[18] LEVOIN N,VO D D,GAUTIER F,et al.A combination of in silico and SAR studies to identify binding hot spots of Bcl-xL inhibitors[J].Bioorg Med Chem,2015,23(8):1747-1757.
[19] BRUNCKO M,OOST T K,BELLI B A,et al.Studies leading to potent,dual inhibitors of bcl-2 and Bcl-xL[J].J Med Chem,2007,50(4):641-662.
[20] PARK C M,BRUNCKO M,ADICKES J,et al.Discovery of an Orally Bioavailable Small Molecule Inhibitor of Prosurvival B-Cell Lymphoma 2 Proteins[J].J Med Chem,2008,51(21):6902-6915.
[21] TS弗蘭奇克二世,DR希爾,AR海特,等.用于制備凋亡啟動子ABT-263的方法[P].中國專利,102131792,2011-07-20.
[22] 張培權,肖貽崧,張守南,等.4-(4-((2-(4-氯苯基)-5,5-二甲基環己基-1-烯)甲基)哌嗪-1-基)苯甲酸的合成方法[P].中國專利,102584744,2012-07-18.
[23] ZHAI H X,XIONG C.Bcl-2 inhibitors[P].US,2009036035,2009-03-19.
[24] K米勒-莫斯林,B-B托雷,MS維塞爾,等.作為Bcl-2家族蛋白抑制劑用于癌癥的治療的磺酰胺化合物[P].中國專利,102498111,2012-06-13.
[25] P卡薩拉,T勒迪瓜爾赫,O熱內斯特,等.新的三元環化合物、它們的制備方法和含有它們的藥物組合物[P].中國專利,101270120,2008-09-24.
[26] BAELL J B,LESSENE G L,SLEEBS B E,et al.Arylsulfonamide compounds[P].US,2008061208,2008-05-22.
Synthesis of ABT-263 Intermediate Carboxylic Acid
SHENHuanling,ZHOUGuochun
(School of Pharmaceutical Sciences, Nanjing Tech University, Nanjing 210009, China)
As the key intermediate of an oral Bcl-2 family protein inhibitor ABT-263 (1), the synthetic method for 4-(4-((2-(4-chlorophenyl)-5,5-di-methylcyclohex-1-enyl)methyl) piperazin-1-yl) benzoic acid (2) was reported. It was the reaction of 4,4-dimethylcyclohexanone with phosphorus tribromide andN,N-dimethylformamide to form the first intermediate 2-bromo-5,5-dimethyl-cyclohex-1-ene carbaldehyde (3); and then after the reactions of Suzuki coupling, reduction, halogenation, amidation, hydrolysis and acidification to afford (2). It was prepared in 47% total yield by six steps, significantly higher than the reported yield. The synthesis with simple and mild conditions, convenient workup procedure is suitable for large scale production. The structures of all products were confirmed by1H NMR.
ABT-263; carboxylic acid fragment; synthesis
2016-11-18
國家自然科學基金資助項目(30973621)
申煥玲(1989-),女,河北邯鄲人,碩士生,主要從事藥物化學方面的研究,(E-mail)shenhuanling@njtech.edu.cn; 周國春(1964-),男,江蘇儀征人,博士,博士生導師,主要從事藥物化學方面的研究,(E-mail)gczhou@njtech.edu.cn
1673-1549(2017)02-0011-05
10.11863/j.suse.2017.02.03
TQ463+.4
A
