偏頭痛相關的發作性動作誘發性運動障礙
——漢族家系臨床及遺傳學特征
2017-05-15 07:29:56畢光輝曲星華劉增玲劉長玲
中國醫藥指南 2017年1期
關鍵詞:動作
畢光輝 曲星華 劉增玲 劉長玲
(東營市人民醫院神經內科,山東 東營 257091)
偏頭痛相關的發作性動作誘發性運動障礙
——漢族家系臨床及遺傳學特征
畢光輝 曲星華 劉增玲 劉長玲
(東營市人民醫院神經內科,山東 東營 257091)
目的 探討合并偏頭痛漢族家系發作性動作誘發性運動障礙(Paroxysmal kinesigenic dyskinesia,PKD)誘發因素、臨床及遺傳學特點。方法 對1個漢族PKD家系共13名成員進行PRRT2基因檢測,其中患者4例,總結其誘因及臨床遺傳特點、藥物治療效果。結果 該家系4例患者均有偏頭痛,3例患者為男性,1例為女性,男女比3∶1,4例對苯妥英鈉療效顯著,該家系PRRT2基因檢測結果顯示該家系中4例存在突變c.694G>A,符合常染色體顯性遺傳。結論 PKD男性多發,臨床表型具多樣性,除運動外,非運動因素如偏頭痛亦可導致PKD發作。
動作誘發性運動障礙;臨床;遺傳學;誘發因素
發作性運動障礙(Paroxysmal dyskinesias,PxDs)是一組少見的神經系統發作性疾病,其中,發作性動作誘發性運動障礙(PKD)是最常見的發作類型,該病的首個致病基因PRRT2(proline-rich transmembrane protein 2)于2011年被克隆[4]。PRRT2致病突變在其他發作性疾病中也有發現[1]。本文報道的PKD家系除了動作誘發外,偏頭痛亦是其誘發因素,通過對該家系研究,進一步了解其臨床及遺傳學特點。
1 資料與方法
1.1 臨床資料:本家系患者發病年齡13~17歲,男性3例,女性1例,男女比3∶1,發病特點為單側及雙側發作性肌強直,動作及偏頭痛誘發,每次發作持續時間不超過1 min,每日發作1~30次,發作時及發作間期均無意識障礙,臨床輔助檢查正常,4例患者均合并偏頭痛,對抗癲癇藥敏感,特別是苯妥英鈉療效顯著,對妥泰亦有效。
1.2 方法
1.2.1 先證者Ⅲ入院后查血生化,甲功系列,及甲狀旁腺激素,排除糖尿病甲狀腺功能亢進等代謝性疾病,行顱腦MRI、腦電圖、肌電圖、心電圖、頭頸部血管超聲、頭頸部MRA等檢查均未發現異常。患者Ⅰ、Ⅱ1、Ⅱ2未住院院治療,在門診給予上述檢查未見異常。
1.2.2 基因檢測:①外周血基因組DNA提取:經東營市人民醫院倫理委員會研究批準,先證者及家系成員均簽署知情同意書后抽取其外周血,提取全部受檢個體外周血基因組DNA,采用標準酚-氯仿法提取基因組DNA,TE緩沖液溶解,-20 ℃保存備用。②引物合成:利用Primer 3在線設計軟件進行引物設計,涵蓋PRRT2所有外顯子編碼區及內含子-外顯子剪切區。③聚合酶鏈式反應及Sanger測序、比對:采用標準PCR反應體系,于相應的退火溫度條件下進行PCR反應,2%瓊脂糖凝膠電泳進行反應產物的鑒定及純化,ABI3730自動測序儀進行PRRT2測序。測序結果應用DNAStar軟件與標準序列進行比對分析。
2 結 果
2.1 遺傳特征:該家系連續3代發病,符合常染色體顯性遺傳(家系譜圖見圖1)。
2.2 基因檢測結果:該家系成員Ⅰ、Ⅱ1、Ⅱ3、Ⅲ1的PRRT2基因均在3號外顯子存在突變c.694G>A,突變類型為錯義突變,其余成員均未發現基因突變(圖2)。
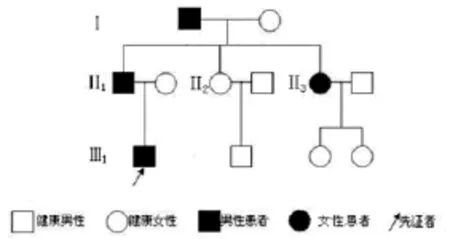
圖1 PKD——家系譜圖

表1 引物序列及反應條件
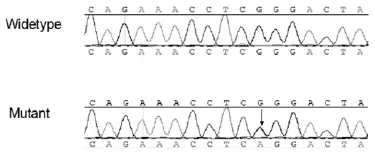
圖2 基因測序圖
3 討 論
發作性運動障礙(Paroxysmal dyskinesias,PxDs)目前根據誘發因素可將其分為4種類型,即發作性運動誘發性運動障礙(paroxysmal kinesigenic dyskinesia,PKD)、發作性過度運動誘發性運動障礙、發作性非運動誘發性運動障礙及發作性夜間睡眠性運動障礙[2]。PKD是最常見的發作類型,2004年Bruno等[3]提出其臨床診斷標準;本文報道的1家系4例患者符合Bruno提出的診斷標準。家族性患者的遺傳方式以常染色體顯性遺傳為主,有的呈不完全外顯,也有呈常染色體隱性遺傳的家系報道[5],本文報道的家系為典型常染色體顯性遺傳,單純PKD家系較為少見,大多伴發其他疾病,本文報道家系即合并偏頭痛。迄今為止,共有3個位點被報道與發作性動作誘發性運動障礙有關,即EKD1~3[6-8]。2011年,來自中國的研究小組率先證實了PRRT2基因為家族性發作性動作誘發性運動障礙的致病基因[4]。本文報道的家系,基因檢測結果PRRT2基因3號外顯子均存在突變c.694G>A,突變類型為錯義突變,再次證實PRRT2為PKD致病基因。系列研究發現PRRT2基因突變也存在于其他一系列發作性疾病,并由此提出了“PRRT2 相關疾病(PRD)”的概念。PRRT2基因包含4個外子,其中第2和3外顯子為主要突變部位,我們研究的PKD家系基因突變都位于PRRT2基因第3外顯子。在PRRT2相關疾病中主要突變類型為無義突變,其他突變類型包括錯義突變、剪切突變和插入突變[6]。c.649dupC(p.R217PfsX8)為熱點突變,因此,對于PKD家系患者,進行PRRT2基因檢測時,c.649dupC(p.R217PfsX8)可列為首選。我們報道的家系致病基因PRRT2,突變c.694G>A,突變類型為錯義突變,也屬較常見的突變類型,一種突變可導致多種表型,PRRT2基因突變的基因型與表型之間的關系目前還無法確定。
目前PKD發病機制尚未明確,PRRT2基因編碼一個跨膜蛋白2,具有重要生理功能。大腦皮質以及小腦、小腦腳、蒼白球、尾狀核、丘腦底核等結構中可檢測到高表達的PRRT2 mRNA[4]。無義突變引起肽鏈截短并介導mRNA降解,導致PRRT2蛋白功能喪失[8],因此PRRT2蛋白功能缺失引起細胞膜定位功能異常,可能與PKD發病有關。此外PKD與突觸相關蛋白25(SNAP25)有關,PRRT2蛋白發生功能缺失后,SNAP25功能受到影響,導致電壓門控性鈣離子通道調控失常,影響突觸囊泡胞吐過程誘發PKD。本研究家系合并偏頭痛,研究發現家族性偏癱性偏頭痛(familial hemiplegic migraine,FHM)是一種與遺傳因素明顯相關的先兆性偏頭痛。患者發病年齡一般在兒童期或青少年期。2012年Dale等[9]在伴有偏癱性偏頭痛的發作性運動障礙家系中檢測出PRRT2基因C.649dupC突變,此后Gardiner等[10]研究發現PRRT2在偏癱性偏頭痛家系中檢出率6.3%(4/49),散發患者為1.1%(2/180),這證明PRRT2是家族性偏癱性偏頭痛致病基因之一。本研究所獲得致病基因是否也導致了該家系偏頭痛,或者偏頭痛是一臨床表型,抑或是PKD的另一誘發因素,PKD是否還存在更多的誘發因素,有待于進一步探討。
[1] Wood H.Genetics:expanding the spectrum of neurological disorders associated with PRRT2 mutations [J].Nat Rev Neurol,2012,8(12): 657.
[2] Demirkiran M,Jankovic J.Paroxysmal dyskinesias;clinical features and classification[J].Ann Neurol,1995,38:571-579.
[3] Bruno MK,Hallett M,Gwinn-Hardy K,et al.Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria[J].Neurology,2004,63(12):2280-2287.
[4] Chen WJ,Lin Y,Xiong ZQ,et al.Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia[J].Nat Genet,2011,43(12):1252-1255.
[5] 周瑾瑕,李國良,劉鼎,等.家族性發作性運動誘發性運動障礙六個家系的臨床及遺傳特點分析[J].中華神經科雜志,2006,39(11): 726-729.
[6] Lee HY,Huang Y,Bruneau N,et al.Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions [J].Cell Rep,2012,1(1):2-12.
[7] Spacey SD,Valente EM,Wali GM,et al.Genetic and clinical heterogeneity in paroxysmal kinesigenic dyskinesia: evidence for a third EKD gene[J].Mov Disord,2002,17(4):717-725.
[8] Cartegni L,Chew SL,Krainer AR.Listening to silence and understanding nonsense: exonic mutations that affect splicing[J].Nat Rev Genet,2002,3(4):285-298.
[9] Dale RC,GardinerA,Antony J,et a1.Familial PRRT2 mutation with heterogeneous paroxysmal disorders including paroxysmal torticollis and hemiplegic migraine[J].Dev Med Child Neurol,2012,54(10): 958-960.
[10] Gardiner AR,Bhatia KP,Stamelou M,et a1.PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine[J].Neurology,2012,79(21):2115-2121.
Clinical and Genetic Features of One Han Nationality Family with Paroxysmal Kinesigenic Dyskinesia and Migraine
BI Guang-hui, QU Xing-hua, LIU Zeng-ling, LIU Chang-ling
(Department of Neurology, Dongying People's Hospital, Dongying 257091, China)
Objective To study the clinical and genetic features and inducing factors of one Han nationality family with paroxysmal kinesigenic dyskinesia (PKD) and migraine. Methods The information of 13 family members in one pedigree, including 4 patients were analyzed, and the responce to treatment and inducing factors and the clinical and genetic features were followed up. DNA was extracted from peripheral blood samples, and then screened for PRRT2 mutations. Results There were four patients with migraine in the pedigree (three men, one women); the ratio of male to female was 3∶1; the four patients showed good response to phenytoin we detected a nonsense mutation c.694G>A in PRRT2 gene in four family members, showing autosomal dominant inheritance. Conclusions PKD with multiple clinical phenotypes occur in more than man, besides kinesigenic factor nonkinesigenic factors such as migraine induced PKD.
Paroxysmal kinesigenic dyskinesias; Clinical; hereditary; Inducing factors
R747.2
B
1671-8194(2017)01-0013-02
猜你喜歡
作文周刊·小學一年級版(2022年16期)2022-05-07 11:28:30
作文周刊·小學一年級版(2021年8期)2021-07-07 11:00:47
動漫界·幼教365(大班)(2021年4期)2021-05-23 21:33:16
小學生作文(低年級適用)(2018年3期)2018-04-17 00:58:35
少年博覽·小學低年級(2017年4期)2017-06-09 16:22:28
作文周刊·小學一年級版(2016年28期)2017-06-03 00:28:49
作文評點報·低幼版(2017年7期)2017-03-11 20:49:41
少兒科學周刊·少年版(2015年4期)2015-07-07 20:56:37
電影故事(2015年30期)2015-02-27 09:03:12
七彩語文·低年級(2014年10期)2015-01-14 14:46:27
