突觸異常發育在孤獨癥譜系障礙發生機制中的研究進展
2017-06-04 01:59:32陳麗萍吳海濤
中國藥理學與毒理學雜志 2017年12期
關鍵詞:小鼠
陳麗萍,吳海濤
(軍事醫學研究院軍事認知與腦科學研究所,北京 100850)
孤獨癥譜系障礙(autism spectrum disorders,ASD)是一種絕大部分由遺傳因子決定的神經發育性精神疾病。大多數ASD患者均在2~3歲前后被診斷出來,而2~3歲正是人大腦發育過程中突觸形成和活性重塑的重要時期。通過對ASD患者進行磁共振成像(MRI)掃描研究發現,ASD患者存在大腦結構異常,在2~4歲時大腦的容量大小會出現一個高峰,隨年齡的增長到青少年時期降低[1-2],并且在2~4歲前后ASD患兒大腦皮質會出現增厚癥狀[3]。突觸是大腦的基本組成單元,是神經細胞之間進行通訊的基本單位,許多突觸蛋白在此過程中發揮至關重要的生理功能。對突觸進行不同類型的刺激可導致突觸更易被激活或抑制,謂之突觸可塑性(synaptic plasticity),這種可塑性也是大腦進行學習記憶和情感交流等認知功能的重要神經生物學基礎。與ASD相關的致病基因雖目前尚未完全揭示,但在已鑒定出的關聯基因中有一大類重要成員編碼突觸相關蛋白,從而在分子層面進一步揭示了突觸發育和功能異常與ASD發生之間的密切關系。
已有研究結果表明,同ASD譜系障礙密切相關的常見單基因突變有脆性X復合體(fragile X syndrome,FXS)FMR1基因,結節性硬化綜合征(tuberous sclerosis complex,TSC)TSC1和TSC2基因,多發性神經纖維瘤病NF1基因,天使綜合征(angelman syndrome,AS)UBE3A基因,Rett綜合征MECP2基因和染色體22q13.3缺失綜合征SHANK3基因等。分別編碼突觸黏附蛋白神經配蛋白(neuroligins)和神經連接蛋白類(neurexins)的NLGN和NRXN基因突變也會導致ASD的發生。誘發形成ASD譜系障礙的相關基因雖目前尚不能被完全揭示,但在ASD患者中具有高頻突變的基因大多數為突觸功能相關關鍵調控分子,這為理解ASD譜系障礙及相關疾病發生的神經機制和藥物研發提供了重要參考依據。
1 孤獨癥相關突觸分子
1.1 神經連接蛋白與神經配蛋白
神經連接蛋白和神經配蛋白是富集于突觸的細胞黏附分子,在哺乳動物體內存在3種編碼神經連接蛋白的基因亞型,依次是Nrex1/2/3,每個基因可編碼α與β 2種亞型的同源異構體[4]。神經配蛋白是神經連接蛋白的內源性配體,在人體內編碼NLGN1/2/3/4共4個基因。通過單個位點可變剪接產生NLGN2/3/4,2個位點可變剪接產生NLGN1。
神經連接蛋白是存在于突觸前膜的單次跨膜蛋白,可與突觸后膜的單次跨膜黏附分子神經配蛋白相互作用。在突觸前膜,神經連接蛋白可與鈣/鈣調蛋白依賴性絲氨酸蛋白激酶(calcium/calmodulindependent serine protein kinase,CASK)在胞內相互結合,而神經配蛋白則與突觸后密度蛋白95(postsynaptic density protein 95,PSD95)在突觸后膜的致密區相互結合[5]。不管是興奮性突觸還是抑制性突觸,突觸前后膜之間的相互作用在突觸分化與成熟中都起著至關重要的作用[5-6]。NLGN基因突變可降低其在突觸后膜致密區的聚集并降低其與神經連接蛋白結合的親和力。在小鼠中,對神經連接蛋白與神經配蛋白2者中任一基因進行敲除均會導致突觸傳遞功能缺陷。此外,許多實驗均證實,神經配蛋白與神經連接蛋白之間相互作用在突觸發育與遞質釋放中均發揮關鍵調控作用。
存在于X染色體上的NLGN3和NLGN4基因是第一批被鑒定出來的能引起ASD的突變基因。在一對同胞兄弟中,其中一個男孩NLGN3基因的一段高保守區域錯義突變引起編碼蛋白產物R451C突變,從而出現了ASD、嚴重智力缺陷和癲癇等癥狀;而其兄弟則患上了阿斯伯格綜合征(Asperger syndrome)。而他們的母親則是NLGN3突變攜帶者,卻無任何臨床癥狀,提示這是一種典型的X染色體相關的遺傳模式[7]。NLGN4突變是在一個男孩ASD患兒身上發現的,NLGN4基因突變引起移碼突變和編碼蛋白的提前終止,其兄弟患上了阿斯伯格綜合征,而母親是一名健康的突變攜帶者[7]。其他有關NLGN4的移碼突變是在一個存在與X染色體相關智力障礙患者的大家族成員和一對同胞兄弟中發現的。在這2兄弟中,一個表現出ASD和智力障礙等癥狀,另一個則出現多動癥、認知障礙和Tourette綜合征等癥狀[8-9]。由此可見,NLGN3和NLGN4基因突變可引起一系列社交和認知障礙,從而導致包括ASD及其他嚴重認知缺陷在內精神疾病的發生。
首次在ASD患者中鑒定出NRXN基因突變的現象是在2名自閉癥患者中發現NRXN1基因及其所在染色體異常[10]。隨后,人們在一名患有生理和認知缺陷以及ASD傾向的男孩身上發現NRXN1基因片段的缺失,這段基因片段能編碼α啟動子和外顯子1~5區域,該片段的缺失導致了下游β啟動子無法正常發揮功能[11]。2009年,一名具有ASD傾向,同時伴有嚴重智力障礙、呼吸過頻和部分發育畸形的患者體內也檢測出NRXN1基因出現缺失突變和翻譯提前終止突變的混合型雜合突變[12]。此外,在精神分裂癥患者中也存在一些NRXN1基因的缺失及重復突變。關于NRXN2基因,人們也在一名ASD患者及其患有語言遲緩癥的父親體內發現了NRXN2基因的一個移碼突變[13]。這些珍貴的臨床資料表明,NRXN基因與ASD、智力障礙以及精神分裂癥等相關疾病均具有非常密切的聯系。
1.2 Shank蛋白
Shank蛋白是位于突觸后膜致密區的一大類突觸后框架蛋白,其中包括與鳥苷酸激酶相關蛋白(PSD95結合蛋白或SAPAP)相互作用的Shank1/2/3、Homer、皮動蛋白結合蛋白以及生長抑素受體。Shank蛋白還可與突觸后膜上的神經配蛋白產生直接或間接相互作用,與PSD95、SAPAP、谷氨酸受體以及細胞骨架形成一個巨大蛋白復合體。所有Shank蛋白都能在大腦中表達,尤其是在皮質和海馬神經元中。但不同的SHANK基因在大腦不同區域的表達模式和表達量存在差別[14]。Shank蛋白可與突觸后密度蛋白相互作用并穩定PDS-95/SAPAP/Shank/Homer復合體。另外,Shank蛋白可將肌醇三磷酸受體和纖絲狀肌動蛋白募集到突觸后膜,從而擴大和穩定樹突棘末端,因此,Shank蛋白是參與調控突觸棘形態的重要分子。
在小鼠海馬神經元中過表達Shank1蛋白能引起樹突棘增大和成熟度增加[15],而敲除Shank1則會出現樹突棘變小、PSD變薄和突觸傳遞降低。并且,Shank1缺失小鼠會呈現焦慮樣行為,異常語境恐怖記憶以及在空間任務中長期停留行為的減少[16]。Shank3+/-雜合突變小鼠也可導致海馬CA1區神經元基礎水平突觸傳遞和長時程增強(longterm potentiation,LTP)的降低。除了突觸功能異常外,Shank3+/-雜合雄性小鼠識別雌性小鼠的社交能力也有所降低[17]。并且,在Shank3B-/-小鼠紋狀體中,突觸后膜上的改變還包括興奮性突觸傳遞的降低。Shank3+/-和Shank3B-/-2種模型小鼠的研究結果均表明,在Shank3突變小鼠中谷氨酸能信號和α-氨基-3-羥基-5-甲基-4-異噁唑丙酸(α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid,AMPA)受體介導的突觸傳遞出現了紊亂,增強AMPA傳遞可能會有助于Shank3單倍劑量不足而引發的相關疾病的治療。最新研究表明,Shank3突變ASD模型小鼠中,紋狀體D2型多棘神經元形態和功能改變對小鼠重復刻板行為有重要影響[18]。
Phelan-McDermid綜合征(PMS)是一種由22號染色體q13位出現一個微小的基因缺失引起的新生兒重癥肌無力,體態異常以及全身發育遲緩的疾病。因此,人們希望從22號染色體上鑒定出PMS致病基因。后來發現,具有SH3和錨蛋白結構域的SHANK3基因位于此位置上,且具有許多神經系統的表現。2001年,Bonaglia[19]首次在一名兒童患者鑒定出SHANK3基因發生易位突變。隨后,研究者通過從60多名患者提取DNA進行分析鑒定發現,SHANK3基因的雜合性丟失是導致神經系統表型的罪魁禍首。研究者在PMS疾病研究中發現,促使SHANK3基因在其他神經精神疾病致病中起重要作用,其中就包括ASD。2007年,在患有ASD和智力障礙的患者體內均發現了SHANK3基因缺失引起的蛋白截斷突變[20-21]。此外,在ASD患者中還發現了SHANK2基因的缺失和終止突變[22],從而進一步證實了SHANK家族基因突變在ASD發生過程中的重要意義和價值。
1.3 UBE3A
包括泛素化在內的蛋白質翻譯后修飾是一類參與調控突觸許多生理過程的重要機制。其中,UBE3A基因編碼一種E3泛素連接酶,能將泛素鏈連接到靶蛋白并標記上,使得靶蛋白能夠順利地被蛋白酶體系統識別降解。研究發現,敲除UBE3A基因可引起小鼠大腦海馬神經元出現嚴重LTP紊亂和學習記憶能力的降低[23]。此外,UBE3A還能夠調控皮質可塑性,其中包括參與調控抑制性中間神經元突觸前膜囊泡循環以及興奮性谷氨酸能神經遞質的釋放等[24-26]。
由于UBE3A可直接通過調控蛋白泛素化修飾調節突觸中靶蛋白的含量,因此UBE3A水平降低引起的突觸功能和可塑性異常可能與其靶蛋白在突觸中的異常堆積有關。在UBE3A眾多靶蛋白中,活性調控細胞骨架相關蛋白(cytoskeletonassociated protein,Arc)是其中一個關鍵靶分子[27]。Arc可通過調控AMPA受體內吞從而對突觸可塑性發揮重要調節作用。通過學習記憶等行為經歷的獲得可提高神經活性和UBE3A表達水平,并可通過UBE3A泛素化降解Arc的方式發揮負反饋調節作用。UBE3A缺失可導致Arc表達上升,從而誘發AMPA受體內吞和興奮性突觸傳遞異常[28]。在母系UBE3A缺失的小鼠中,Arc表達量會隨著突觸活性的提高而增加;在結構水平上,UBE3A缺失可導致樹突棘發育紊亂,包括樹突棘的形態、數量以及長度[29]。通過對錐體神經元高度極化的樹突形態進行深入研究證實,UBE3A能夠調控樹突的生長和極性。此外,在UBE3A突變小鼠中,人們還發現鈣調素依賴蛋白激酶Ⅱ(Ca/calmodulin-dependent protein kinaseⅡ,αCaMKⅡ)中一個抑制性磷酸化位點突變能夠挽救UBE3A突變引起的LTP和學習記憶缺陷[30]。
最近一項關于UBE3A雜合突變小鼠誘發AS的小鼠模型研究顯示,UBE3A雜合突變小鼠海馬區中代謝型谷氨酸受體5(metabotropic glutamate receptor,mGluR5)介導的長時程抑制(long-term depression,LTD)會出現選擇性擴增[31]。但這種改變并不是由于下游信號通路改變或者Arc表達水平升高引起的,而是與突觸后膜蛋白Homer1a亞型表達量降低和mGluR5受體與Homer1b/c亞型結合增加密切相關。mGluR5介導的LTD過度增強可以被mGluR5拮抗劑所糾正。此外,在UBE3A突變的AS模型小鼠中,海馬錐體神經元中鈉/鉀-三磷酸腺苷酶 α1亞 基(α1-sodium-potassium ATPase,α1-Na+/K+-ATPase)和軸突起始段蛋白表達量增加[32]。這些改變可能會影響神經元興奮性,并改變大腦海馬區依賴的學習記憶能力,通過對神經元的α1-Na+/K+-ATPase進行遺傳敲減能夠預防AS小鼠中神經元軸突起始段異常,改善突觸LTP和海馬相關的學習記憶缺陷。
1.4 脆性X智力低下蛋白
脆性X智力低下蛋白(fragile X mental retardation protein,FMRP)是一類在神經元中高表達,并且能夠在神經元胞體和樹突中與mRNAs相互結合的蛋白。目前,FMRP功能研究有一個比較引人注目的發現,就是FMRP在突觸后膜能夠選擇性地與mRNA結合,從而負調控一系列樹突mRNA的轉錄[33]。FMRP的靶蛋白包含了突觸傳遞中的大量蛋白,且其中許多蛋白是ASD研究中的候選靶基因產物。FMRP蛋白缺失可引起樹突蛋白的過量合成。另外,FMRP還能通過調控突觸前膜神經遞質釋放來控制海馬錐體神經元動作電位的持續時間[34]。因此,FMRP缺失能夠導致動作電位持續時間的延長,增強突觸前膜鈣離子內流和重復激活狀態下神經遞質的釋放。與突觸后膜不同,這種突觸前的作用機制并不依賴于蛋白翻譯,而是由大電導鈣離子激活鉀離子通道所介導的[34]。
FMRP在突觸中的具體作用機制目前認為主要與mGluR介導的信號有關。FMRP在突觸接受刺激后通過抑制突觸后膜mRNA蛋白翻譯從而對mGluR依賴的LTD發揮調控[35-36]。mGluR介導的LTD失調是引發FXS患者中許多神經系統病癥的原因之一,在FMR1敲除小鼠中抑制mGluR5能夠部分恢復突觸中蛋白合成水平[37-38]。在一種ASD表型的BTBR模型小鼠中,部分抑制mGluR5亦能顯著改善其異常行為癥狀[39]。FMRP的缺失除了會影響mGluR相關的突觸可塑性以外,還會影響多巴能和γ-氨基丁酸(gamma-aminobutyric acid,GABA)能信號。在FMR1基因敲除小鼠和FXS模型果蠅中均出現了GABA-A受體表達量的顯著降低[40]。此外,在FMR1基因缺陷小鼠中,多巴胺依賴的LTP減弱,給予多巴胺拮抗劑可以顯著提高FXS患者的活力和運動癥狀[41-42]。
在FMR1缺失小鼠及來源于FXS患者的成纖維細胞中,哺乳動物雷帕霉素靶蛋白(mammalian target of Rapamycin,mTOR)信號通路顯著激活,mTOR激酶活性及其下游靶蛋白S6激酶和4E-BP磷酸化水平均顯著提高,提示FMRP可負調控mTOR信號通路活化[43]。在FMR1缺失小鼠中,胞外信號調節激酶(extracellular signal-regulated kinase,ERK)信號增加還會直接影響蛋白的翻譯速率。并且,羥甲基戊二酰輔酶A還原酶(hydroxy methylglutaryl coenzyme A reductase inhibitor,HMG-CoA)抑制劑和ERK1/2信號通路的阻斷劑洛伐他汀(lovastatin)能減輕該模型小鼠的許多缺陷表型[44]。糖原合酶激酶 3β(glycogen synthase kinase 3β,GSK3β)是FMRP另一個靶蛋白,其活性在FXS果蠅和小鼠中顯著增加,采用GSK3β抑制劑鋰處理FXS果蠅和小鼠可顯著改善其分子和行為學表型。在另一項開放式研究中,也證實了鋰可適度提高和改善FXS患者的異常行為問題[45]。上述研究結果表明,能夠作用于mTOR或ERK信號通路及其下游靶蛋白(如磷脂酰肌醇-3-羥激酶)的抑制劑可作為糾正FXS患者蛋白合成和突觸相關缺陷的潛在候選目標。
1.5 TSC1和TSC2
蛋白harmarin(TSC1)和tuberin(TSC2)分別由TSC1和TSC2基因編碼,TSC1與TSC2蛋白可與TBC1結構域家族成員7(TBC1 domain family member 7,TBC1D7)蛋白三者相互結合形成蛋白復合物。這種蛋白復合物作為小G蛋白RheB(在腦中富集的Ras同源物)的GTP酶激活蛋白,能響應細胞內多種信號。TSC1/2蛋白復合物對神經元樹突棘生成和軸突生長非常重要,TSC1或TSC2缺失可導致樹突棘結構和密度紊亂[46]以及軸突導向受損[47]。TSC2+/-雜合突變小鼠可出現mTOR信號通路過度激活和自噬障礙。此外,研究者從散發性ASD患者死后的大腦顳葉組織中發現樹突棘密度增大和成熟缺陷,這可能與TSC-mTOR信號通路相關[48]。
TSC是由TSC1和TSC2基因突變引發的遺傳性多系統神經皮膚綜合征。在30%家族性遺傳患者和15%散發性患者體內均發現TSC1基因的突變,而更高比例的家族性遺傳患者和50%散發性患者體內則出現TSC2基因的突變。TSC患者中許多中樞神經系統癥狀的病理生理學機制正在逐漸被揭示,包括細胞增殖和遷移失調、軸突生長改變、突觸生成和髓鞘化受損等,以及由此引發的中樞神經系統解剖結構的破壞和神經連接的改變等[49]。TSC轉基因小鼠模型能很好地模擬ASD患者的核心癥狀,非常適合用來研究ASD大腦神經連接異常的機制。在小鼠小腦的浦肯野細胞中特異性地敲除TSC1或TSC2基因能引發小鼠出現社交障礙、刻板行為和異常發聲等ASD樣表型[50-51]。組成型的TSC1或TSC2雜合型基因敲除小鼠也會出現不同程度的認知和社交障礙[51-52]。
利用免疫抑制劑西羅莫司(雷帕霉素,sirolimus,Rapamycin)對TSC突變小鼠進行藥物處理可減輕小鼠的一些行為學缺陷表型。西羅莫司是mTOR信號通路抑制劑。此外,對成年小鼠進行西羅莫司處理可糾正TSC突變小鼠中晚期LTP表型并改善其學習記憶缺陷[52]。這表明TSC患者中的一些突觸和行為表型與FXS和Rett綜合征相似,即都可能是由于持續性病理生理過程所引發,而不是發育過程中一個不可逆的突發事件導致的,這些病理生理學變化可望通過藥物加以糾正。
1.6 甲基CpG結合蛋白2
甲基CpG結合蛋白2(methyl CpG binding protein 2,MECP2)是一種存在于細胞核內的甲基化胞嘧啶結合蛋白,屬于甲基化CpG結合蛋白家族成員。MECP2基因位于X染色體q28上。研究表明,MECP2可通過與組蛋白去乙酰化酶(histone deacetylase,HDAC)復合物相互作用發揮轉錄抑制功能[53-54]。意外的是,某些基因轉錄在MECP2缺失條件下會被抑制,而在其激活條件下出現過表達,表明MECP2不是一種典型的轉錄抑制子。MECP2可結合到這些靶基因的啟動子區域,這些被MECP2正調控的基因中就包括關鍵的腦源性神經營養因子(brain derived neurotrophic factor,BDNF)等。除了與靶基因進行特異性結合外,染色質免疫共沉淀(chromatin immunoprecipitation,ChIP)結果顯示,MECP2在全基因組能廣泛地與DNA 結合[55-56]。
MECP2在神經元表達水平很高,并且在動物出生后隨神經元成熟蛋白水平會進一步增加。最近的研究結果顯示,在神經膠質細胞中也有MECP2的表達。雖其在膠質細胞中的表達較神經元低,但MECP2在膠質細胞中的缺失可導致MECP2野生型或敲除型神經元樹突形態發育異常,提示MECP2可通過膠質細胞對神經元發育間接發揮調控作用[57]。MECP2缺失的膠質細胞雖可能會引起或加重某些神經系統癥狀,但還有一些研究表明,在神經元中特異性地敲除MECP2足以引起突變小鼠神經系統的功能障礙。
位于X染色體上的MECP2基因突變是引發Rett綜合征的主要病因[58]。絕大部分(99%)Rett綜合征散發病例都有其相應的遺傳基礎,其中97%的典型病例都與MECP2基因的突變有關[59]。MECP2突變引發Rett綜合征的發現揭示這個基因的突變可能導致神經系統功能障礙并出現精神疾病癥狀。除了突變外,MECP2基因的2倍或3倍擴增均能引起進行性神經綜合征,這表明神經系統對MECP2蛋白表達水平的精準度非常敏感[60]。MECP2基因在Xq28片段上出現的2倍或3倍擴增可將多個不同基因共軛在一起,提示MECP2可能是調控ASD發生的關鍵致病分子[61]。MECP2基因異常擴增的男性患者通常出現張力減退和發育遲緩,早期出現ASD癥狀進一步發展為嚴重的運動和認知障礙、癲癇、震顫、Rett綜合征樣癥狀和過早死亡[62-63]。根據X染色體失活模式的不同,女性患者的臨床癥狀主要包括焦慮、抑郁、廣譜ASD表型以及Rett綜合征等[63-64]。因此,MECP2的缺失突變或異常擴增2種不同變化所導致的神經精神系統障礙和異常表型為進一步探索MECP2的調控機制提供了巨大機遇和挑戰。我國學者在構建MECP2基因非人靈長類動物模型方面取得一系列重要標志性成果,其中包括首次在世界上成功建立了MECP2轉基因“ASD”食蟹猴[65],以及利用轉錄激活因子樣效應物核酸酶(transcription activatorlike effector nucleases,TALEN)技術構建的食蟹猴Rett綜合征動物模型[66]。這些成果為深入揭示MECP2基因致病機制以及干預治療等提供了寶貴的研究對象。
1.7 其他突觸相關蛋白
除上述提到的ASD相關突觸分子外,高表達在突觸位置的L型壓力門控鈣離子通道和鈣黏蛋白(cadherin)功能異常也與ASD發生密切相關。患有心肌病的Timothy綜合征患者存在L型鈣離子通道蛋白G406R突變,此類患者會表現出部分典型的ASD癥狀[67]。L型鈣離子通道也存在于神經元的樹突棘和樹突干上,能調控突觸后膜的LTP和突觸可塑性[68]。在突觸間隙中,鈣黏蛋白可通過同源結合將突觸前后膜連接起來,只能促進突觸分化和成熟,并調控突觸可塑性[69]。在部分ASD患者體內發現了編碼鈣黏蛋白的CDH9,CDH10和CDH15基因突變或染色體異常,這些突變會降低突觸連接的穩定性,破壞突觸發生和分化,從而導致ASD的產生。隨著二代高通量測序技術的不斷發展和研究的逐步深入,越來越多的ASD致病基因正在逐漸被發現和報道,通過采用大數據手段對這些數據進行深度挖掘和分析,必將會進一步加深對ASD致病機制的理解。
2 孤獨癥相關重要信號通路
每一個被鑒定出來的ASD易感基因都會為研究ASD發生神經機制提供新的曙光。許多ASD相關基因能歸納到一些主要的信號通路中,即基因轉錄調控與染色質重塑、蛋白合成與細胞代謝以及突觸發育與功能三大方面。有許多細胞過程在神經元和非神經細胞中雖然共性存在,但這些過程在大腦中似乎起著一些與ASD特異性相關的作用(圖1)。
2.1 基因轉錄調控與染色質重塑
De Rubeis等[70]通過采用大規模外顯子組測序方法對上千類ASD患者及其親代進行了深入研究。結果表明,有>5%的ASD患者可檢出新發功能喪失突變,相關基因基因多集中在突觸、轉錄和染色質重塑中發揮功能。如前所述,引起Rett綜合征的MECP2基因就是一個典型的參與多任務、可通過影響染色質重塑、靶基因轉錄和剪輯來調控基因轉錄的關鍵分子。MECP2通過與基因啟動子區甲基化CpG位點結合,或與染色質沉默復合物相互作用來抑制基因表達。也有研究表明,MECP2能與染色質和轉錄激活子相互作用激活蛋白的表達[71]。另外,與FMRP類似的是,MECP2蛋白也可通過轉錄后調控方式參與微小RNA介導的基因表達[72]。
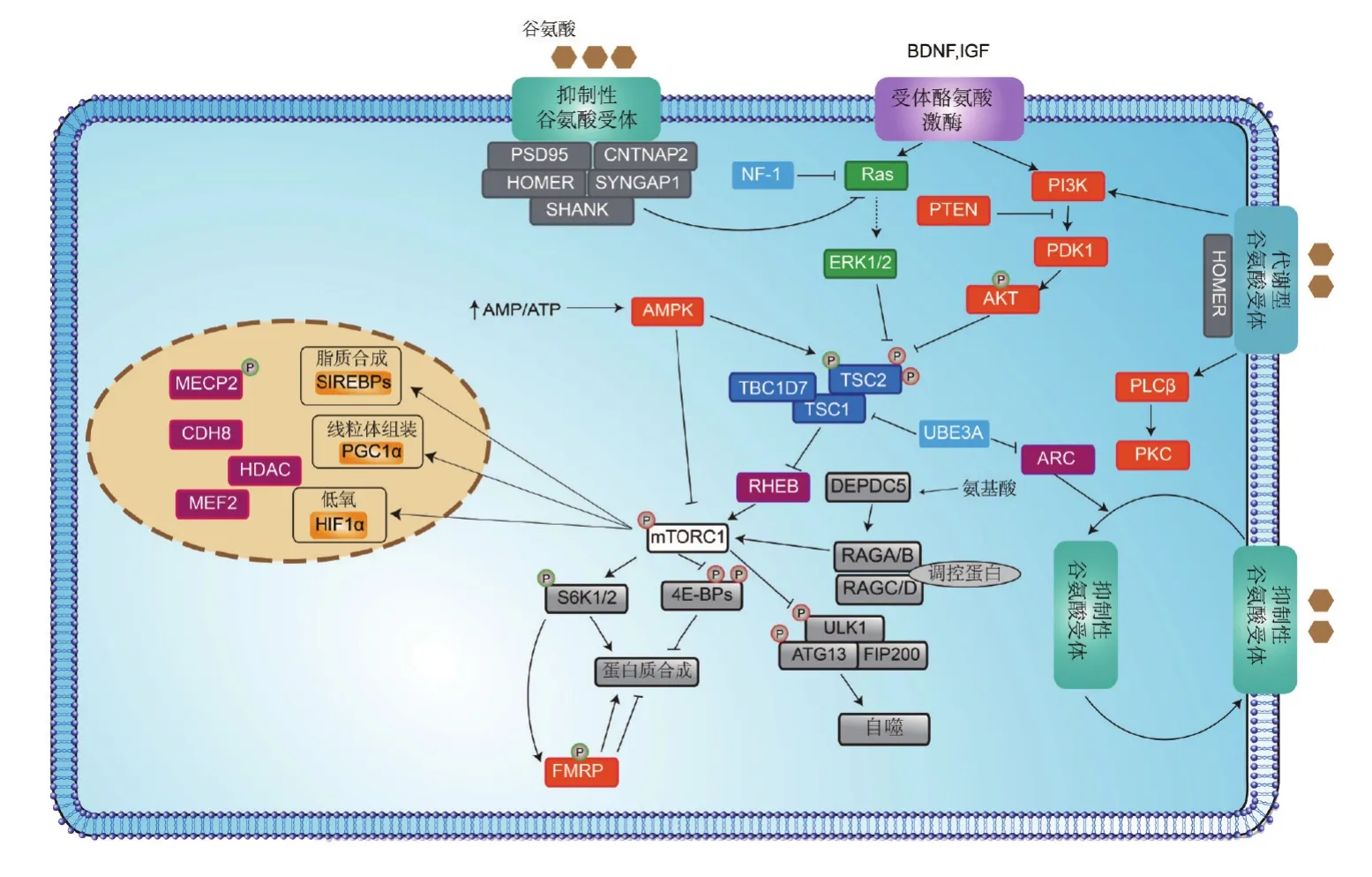
圖1 孤獨癥及相關疾病中突變蛋白所參與的主要信號通路.BDNF:腦源性神經營養因子;IGF:胰島素樣類增長因子;MECP2:甲基CpG結合蛋白2;CDH8:鈣黏蛋白8;HDAC:組蛋白去乙酰化酶;MEF2:肌細胞增強因子2;mTORC1:哺乳動物西羅莫司(雷帕霉素)靶蛋白1;AMPK:腺苷酸活化蛋白激酶;FMRP:脆性X智力低下蛋白;AMP/ATP:磷酸腺苷/三磷酸腺苷;Ras:大鼠肉瘤;RHEB:Ras蛋白腦組織同源類似物;ARC:細胞骨架活性調節蛋白;PTEN:磷酸酶和張力蛋白同源物一種抑癌基因;PI3K:磷脂酰肌醇3激酶;AKT:蛋白激酶B;PDK1:3-磷酸肌醇依賴性蛋白激酶1;PLCβ:磷脂酶Cβ;PKC:蛋白激酶C;ERK1/2:細胞外信號調節激酶1和2;NF-1:神經纖維瘤病1型;UBE3A:15號染色體上UBE3A基因控制合成的蛋白;S6K1/2:核糖體蛋白S6激酶1和2.
此外,最新研究顯示,表觀遺傳修飾也會影響ASD疾病及大腦早期發育調控進程。研究發現一個新的ASD致病基因Auts2基因可通過募集多梳群蛋白(polycomb group proteins,PcG)家族成員胞質分裂調節蛋白1(protein regulator of cytokinesis 1,PRC1)等形成復合體,進而將PRC原有的基因抑制功能逆轉成促轉錄激活因子,從而對大腦發育相關關鍵基因和ASD等疾病相關基因的表達發揮重要調控作用,這也為開發此類疾病新療法提供了新依據[73]。
染色體結構域解旋酶DNA結合蛋白8(chromodomain helicase DNA binding protein 8,CHD8)是一個調控染色質重塑的重要分子,其編碼基因CHD8變異與ASD發生亦密切相關。研究表明,CHD8突變的雜合小鼠胚胎的神經發育出現延遲,成年后可出現類似于ASD患者樣的行為表型,其大腦基因表達在一定范圍內出現了全局性變化。基因集合富集分析顯示,CHD8突變小鼠和人類ASD患者腦內RE-1沉默轉錄因子(RE1-silencing transcription factor,REST)目標基因的表達均有所降低,提示CHD8突變或單倍劑量不足所導致的ASD病理生理學改變可能與REST蛋白的非正常活動及基因異常表達有關[74]。除此以外,包括人第10號染色體缺失的磷酸酶和張力蛋白同源物(phosphatase and tensin homolog,PTEN),β-連環蛋白(βcatenin)等在內的很多分子都可通過調控關鍵靶基因轉錄的方式發揮作用。
2.2 蛋白合成與細胞代謝信號通路
在神經元中,磷脂酰肌醇-3-羥激酶(phosphoinositide 3-kinase,PI3K)/mTOR和GTP結合蛋白Ras(rat sarcoma,Ras)/絲裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)2條蛋白合成和細胞代謝信號通路對突觸功能起到至關重要調節作用,這些信號通路的異常與神經發育和突觸功能障礙密切相關。TSC和PTEN錯構瘤綜合征(PTEN hamartoma tumor syndrome,PHTS)是2種典型的mTOR信號通路介導的疾病,TSC1/TSC2缺失或PTEN功能異常會引起mTOR激酶的激活進而導致智力障礙、癲癇發作以及ASD的發病率增加[42]。在mTOR信號通路中能引起ASD的其他基因有1型神經纖維瘤蛋白(neurofibromin 1,NF1)基因。NF1基因編碼一種可以抑制原癌基因Ras活性并改變mTOR信號通路的三磷酸酯鳥苷激活蛋白,該基因突變會導致Ⅰ型神經纖維瘤疾病。
蛋白合成失調是部分神經發育障礙性疾病的典型特征之一,如脆性X綜合征。FMRP是一類mRNA結合蛋白,參與mRNA翻譯調控并在脆性X綜合征患者中表達沉默,FMRP沉默會導致突觸可塑性相關重要功能分子的蛋白合成出現紊亂。與此類似的是,MECP2也可影響幾百個基因的表達。在MECP2基因突變小鼠中,BDNF和IGF1的表達水平均顯著降低,兩者表達水平的下降可與其他分子協同作用從而導致PI3K/mTOR和ERK/MAPK信號通路活化減少,而給予重組人源IGF1處理能顯著上調這兩條信號通路活化水平[71]。由于PI3K/mTOR和ERK/MAPK信號通路參與調控大量的細胞生物學過程,包括轉錄、自噬、代謝以及細胞器生成和維持等,可見,要深入理解這些信號通路及其所參與的細胞生物學過程在ASD發生機制和治療方案中的意義和價值,仍有待進一步深入探索。
3 孤獨癥相關腦區與神經環路
眾所周知,大腦神經元內分子信號通路的變化往往通過影響神經元和突觸功能,進而影響神經元之間的連接與功能環路,最終導致大腦整體功能的改變。引發ASD的致病機制中有關大腦區域與神經環路的精確信息迄今尚不明了。由于ASD患者伴有多種認知與行為學改變,因此很難將引發ASD的神經機制歸結在某一特定細胞類型或神經環路上。此外,目前已知有上百種基因參與ASD及相關疾病的發生,這些分布在不同節點上的不同基因所參與的細胞類型和神經環路的多樣性也使得對ASD神經環路機制的揭示難度進一步增加。
基于遺傳性ASD模型小鼠的研究表明,在大腦內某些特定區域及一些特定細胞類型存在功能異常。研究還發現,人腦中一些ASD致病基因相互之間存在共表達范式[75-76]。其中一項研究表明,ASD模型鼠大腦中正在形成皮質第5/6質的皮質投射神經元出現富集;還有研究發現,ASD模型鼠大腦中出現淺表皮質和谷氨酸能投射神經元的富集。盡管這2項研究發現的ASD相關大腦皮質的部位并不完全相同,但2項研究均證實,皮質投射神經元異常可能與ASD的發生密切相關且至關重要。
除皮質投射神經元外,研究還發現,其他亞型神經細胞在ASD病理發生中都能起到一定作用。在大量ASD模型小鼠的新皮質內都出現了小清蛋白(parvalbumin,PV)陽性抑制性中間神經元密度的降低[77]。PV特異性地敲除小鼠會出現與ASD患者類似的核心癥狀及行為學表型。與此不同的是,其他研究發現,ASD患者大腦內海馬CA1和CA3區域出現了PV陽性和鈣視網膜蛋白陽性中間神經元的選擇性增加[78]。總之,在某一特定類型細胞和腦區內進行ASD基因的特異性敲除,對解析ASD病理環路及其不同癥候群之間的對應關系至關重要。如前所述,NLGN1敲除小鼠表現出ASD樣重復行為和皮質紋狀體突觸異常。NLGN3突變體具有類似的異常表型,但這些缺陷更像是伏隔核/腹側紋狀體特異性突觸異常引起的[79]。Shank3能在基底神經節中表達,而且Shank3敲除小鼠表現出重復的修飾整理行為、社交異常和紋狀體突觸的改變[18,80]。上述結果表明,基底神經節與皮質的連接異常可能從某些方面部分解釋ASD發生的致病機制。
通過對ASD患者進行組織病理學、影像學及相關損傷的流行病學研究還發現,小腦在ASD病理發生過程中也可能發揮調控作用。神經病理學研究表明,與小腦發育正常的對照組個體相比,小腦浦肯野細胞缺失的個體更易患ASD。此外,對ASD患者進行影像學分析發現,直到兒童期早期患者小腦中還會出現灰質和白質的異常。通過對患有TSC的ASD患者進行正電子成像分析發現,患者的小腦核代謝水平顯著提高,提示小腦皮質的能量輸出增加,而不伴有ASD的TSC患者卻檢測不到此現象[81]。通過對近1000名ASD和正常人的大腦進行全腦關聯分析后,找到了20個與ASD最為相關的腦區,它們包含了多個神經環路,其中一個重要神經環路“顳葉視覺皮質-腹內側前額葉皮質”的功能連接顯著減弱,為深入揭示ASD發生的相關腦區和神經環路提供了重要依據和線索[82]。
4 孤獨癥治療的臨床藥理學研究進展
目前為止,能夠緩解ASD相關癥狀的治療方案仍較為有限。雖然相關研究為ASD治療新方案的研發提供了希望,但從近期一項系統性調查結果看,許多進行藥物治療的ASD患兒并未獲得預期的治療效果[83]。ASD有效治療方案的獲得依然充滿挑戰。如前所述,ASD表型在遺傳、環境、認知和社交等方面的異質性給研究者帶來了諸多困難,這些背景各異的患者使得一種藥物能產生廣泛有效的可能大為降低[84]。另一些阻礙獲得有效治療方案的難度因素還包括樣本量不足、缺乏嚴重受損患者參與、以及缺乏統一評價標準等。此外,不同文化之間的差異性也為ASD人群治療方案的獲得增加了不確定性。
當前認為,治療ASD相關行為異常的黃金治療方案是在患病早期進行一種密集傳遞模式的行為介入。且早期的精細行為學干擾實施起來非常昂貴,需要綜合其他資源方能達到一定療效,這對很多ASD兒童以及家庭來說都是難以承受的。盡管圍繞ASD在基礎神經科學和人類遺傳學研究方面迄今為止已取得諸多進展,但被美國食品藥品監督管理局(FDA)批準用于臨床ASD治療的藥物僅限于利培酮(利哌立酮,risperidone)一種多巴胺拮抗劑)和阿立哌唑(aripiprazole,一種多巴胺受體激動劑)。利培酮是2006年被批準用于成年人的精神抑制藥,可用于治療ASD兒童和青少年出現的易怒和興奮性過強,包括侵略行為、蓄意自傷和發脾氣等癥狀。利培酮是大腦多巴胺和5-羥色胺受體拮抗劑,其短期療效安全有效,可改善ASD患者刻板重復行為和過度興奮等癥狀[85]。不過,副作用嚴重,包括食欲增強引起的肥胖、嗜睡和腦垂體分泌催乳素水平增加等[86]。2009年,在對阿立哌唑的短期療效和安全性進行評價后,FDA批準此藥用于治療兒童和青少年ASD患者的易怒癥狀。阿立哌唑是第三代非典型性精神抑制藥,是一種多巴胺系統穩定劑,不會引起血清中催乳素水平增加及引發錐體外系癥狀。其主要副作用包括疲勞、嘔吐、食欲增加、嗜睡、震顫和因為攻擊和體質量增加引起的不連續服藥等[87]。
由于ASD具有數目龐大的致病候選基因,其遺傳異質性對治療效果的評估具有重要意義。首先,用提高有效干預措施的方式來進行最大同質化治療分組的選擇,可望優化治療的實驗設計。其次,進一步提高對ASD遺傳異質性生物基礎的正確理解,將有利于精準識別和劃分特征各異的患者亞群。此外,對ASD相關臨床合并癥和心理疾病的調查,包括ASD患者免疫系統異常、線粒體功能障礙、消化系統異常、睡眠障礙、癲癇、抑郁和焦慮等,也是一種獲得ASD異質性特點的有效方式。與此同時,ASD遺傳異質性也為其有效治療方案的開發帶來了巨大的障礙和挑戰。一種治療方案或藥物能治愈所有ASD患者固然是一種非常理想的狀態,但在現實情況下其可能性微乎其微,因為ASD是一種典型的多基因遺傳性疾病,某些ASD相關基因在突觸水平甚至具有截然相反的表型[88]。而針對每一種ASD患者的遺傳因素開發特異性治療方案的策略同樣也充滿了不確定性。
現階段最理想的治療方案就是希望能找出一些用有限藥物就可干擾的一些分子和神經環路的集合。目前,相關研究大多集中在ASD外顯率較高的遺傳性狀上,尤其是單基因突變引發的遺傳性狀(表1)。

表1 遺傳性神經發育障礙臨床試驗.
5 展望
ASD作為一種異常復雜的多基因關聯遺傳性精神疾病,在過去十余年間對其認識取得了飛速發展。隨著高通量測序和遺傳連鎖分析技術的運用,一系列ASD致病基因被陸續發現和報道,其中相當大一部分基因直接或間接地參與了突觸發育與功能調控進程。基于腦成像與功能連接組學等技術的運用,也揭示了多個同ASD密切相關的神經環路功能變化,提示ASD發生的機制可能是以網絡狀和多維度調控的方式進行。
目前,用于改善ASD患者主要癥狀的治療方案和藥物仍極為匱乏,ASD的遺傳異質性是導致其療效欠佳的一個根本原因。在缺乏行為學治療的條件下,單獨藥物治療恐難以達到最佳效果。未來,需要遺傳學家、神經科學家、藥理學家和臨床醫師攜手攻關,一方面,盡可能全面深入地揭示ASD相關的致病基因,研發出新型高效藥物;另一方面,需考慮將藥物治療同新型神經調控、行為學精準干預等技術結合,通過多中心、大隊列聯合研究等方式開展協同攻關,并建立標準化、定量化和統一化的病例資源數據庫。從而為解決ASD這一重大難題和挑戰提供基礎理論支撐和潛在治療前景。
[1]Courchesne E,Carper R,Akshoomoff N.Evidence of brain overgrowth in the first year of life in autism[J].JAMA,2003,290(3):337-344.
[2]Courchesne E,Karns CM,Davis HR,Ziccardi R,Carper RA,Tigue ZD,et al.Unusual brain growth patterns in early life in patients with autistic disorder:an MRI study[J].Neurology,2001,57(2):245-254.
[3]Hardan AY,Muddasani S,Vemulapalli M,Keshavan MS,Minshew NJ.An MRI study of increased cortical thickness in autism[J].Am J Psychiatry,2006,163(7):1290-1292.
[4]Tabuchi K,Südhof TC.Structure and evolution of neurexin genes:insight into the mechanism of alternative splicing[J].Genomics,2002,79(6):849-859.
[5]Dean C,Dresbach T.Neuroligins and neurexins:linking cell adhesion,synapse formation and cognitive function[J].Trends Neurosci,2006,29(1):21-29.
[6]Graf ER,Zhang X,Jin SX,Linhoff MW,Craig AM.Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins[J].Cell,2004,119(7):1013-1026.
[7]Jamain S,Quach H,Betancur C,R?stam M,Colineaux C,Gillberg IC,et al.Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism[J].Nat Genet,2003,34(1):27-29.
[8]Laumonnier F,Bonnet-Brilhault F,Gomot M,Blanc R,David A,Moizard MP,Raynaud M,et al.X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene,a member of the neuroligin family[J].Am J Hum Genet,2004,74(3):552-557.
[9]Lawson-Yuen A,Saldivar JS,Sommer S,Picker J.Familial deletion within NLGN4 associated with autism and Tourette syndrome[J].Eur J Hum Genet,2008,16(5):614-618.
[10]Kim HG,Kishikawa S,Higgins AW,Seong IS,Donovan DJ,Shen Y,et al.Disruption of neurexin 1 associated with autism spectrum disorder[J].Am J Hum Genet,2008,82(1):199-207.
[11]Zahir FR,Baross A,Delaney AD,Eydoux P,Fernandes ND,Pugh T,et al.A patient with vertebral,cognitive and behavioural abnormalities and a de novo deletion of NRXN1alpha[J].J Med Genet,2008,45(4):239-243.
[12]Zweier C,de Jong EK,Zweier M,Orrico A,Ousager LB,Collins AL,et al.CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila[J].Am J Hum Genet,2009,85(5):655-666.
[13]Gauthier J,Siddiqui TJ,Huashan P,Yokomaku D,Hamdan FF,Champagne N,et al.Truncating mutations in NRXN2 and NRXN1 in autism spectrum disordersandschizophrenia [J].Hum Genet,2011,130(4):563-573.
[14]Lim S,Naisbitt S,Yoon J,Hwang JI,Suh PG,Sheng M,et al.Characterization of the Shank family of synaptic proteins.multiple genes,alternative splicing,and differential expression in brain and development[J].J Biol Chem,1999,274(41):29510-29518.
[15]Sala C,Pi?ch V,Wilson NR,Passafaro M,Liu G,Sheng M.Regulation of dendritic spine morphology and synaptic function by Shank and Homer[J].Neuron,2001,31(1):115-130.
[16]Hung AY,Futai K,Sala C,Valtschanoff JG,Ryu J,Woodworth MA,Kidd FL,et al.Smaller dendritic spines,weaker synaptic transmission,but enhanced spatial learning in mice lacking Shank1[J].J Neurosci,2008,28(7):1697-1708.
[17]Bozdagi O,Sakurai T,Papapetrou D,Wang X,Dickstein DL,Takahashi N,et al.Haploin sufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function,social interaction,and social communication[J].Mol Autism,2010,1(1):15.
[18]Wang W,Li C,Chen Q,van der Goes MS,Hawrot J,Yao AY, et al.Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism[J].J Clin Invest,2017,127(5):1978-1990.
[19]Bonaglia MC,Giorda R,Borgatti R,Felisari G,Gagliardi C,Selicorni A,Zuffardi O.Disruption of the Pro-SAP2 gene in at(12;22)(q24.1;q13.3)is associated with the 22q13.3 deletion syndrome[J].Am J Hum Genet,2001,69(2):261-268.
[20]Durand CM,Betancur C,Boeckers TM,Bockmann J,Chaste P,Fanchereau F,et al.Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders[J].Nat Genet,2007,39(1):25-27.
[21]Moessner R,Marshall CR,Sutcliffe JS,Skaug J,Pinto D,Vincent J,et al.Contribution of SHANK3 mutations to autism spectrum disorder[J].Am J Hum Genet,2007,81(6):1289-1297.
[22]Berkel S,Marshall CR,Weiss B,Howe J,Roeth R,Moog U,et al.Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation[J].Nat Genet,2010,42(6):489-491.
[23]Jiang YH,Armstrong D,Albrecht U,Atkins CM,Noebels JL,Eichele G,et al.Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation[J].Neuron,1998,21(4):799-811.
[24]Yashiro K,Riday TT,Condon KH,Roberts AC,Bernardo DR,Prakash R,et al.Ube3a is required for experience-dependent maturation of the neocortex[J].Nat Neurosci,2009,12(6):777-783.
[25]Wallace ML,Burette AC,Weinberg RJ,Philpot BD.Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects[J].Neuron,2012,74(5):793-800.
[26]Hayrapetyan V,Castro S,Sukharnikova T,Yu C,Cao X,Jiang YH,et al.Region-specific impairments in striatal synaptic transmission and impaired instrumental learning in a mouse model of Angelman syndrome[J].Eur J Neurosci,2014,39(6):1018-1025.
[27]Kishino T,Lalande M,Wagstaff J.UBE3A/E6-AP mutations cause Angelman syndrome[J].Nat Genet,1997,15(1):70-73.
[28]Greer PL,Hanayama R,Bloodgood BL,Mardinly AR,Lipton DM,Flavell SW,et al.The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc[J].Cell,2010,140(5):704-716.
[29]Dindot SV,Antalffy BA,Bhattacharjee MB,Beaudet AL.The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus,and maternal deficiency results in abnormal dendritic spine morphology[J].Hum Mol Genet,2008,17(1):111-118.
[30]van Woerden GM,Harris KD,Hojjati MR,Gustin RM,Qiu S,de Avila Freire R,et al.Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKⅡinhibitory phosphorylation[J].Nat Neurosci,2007,10(3):280-282.
[31]Pignatelli M,Piccinin S,Molinaro G,Di Menna L,Riozzi B,Cannella M,et al.Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling Angelman syndrome[J].J Neurosci,2014,34(13):4558-4566.
[32]Kaphzan H,Buffington SA,Jung JI,Rasband MN,Klann E.Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome[J].J Neurosci,2011,31(48):17637-17648.
[33]O′Donnell WT,Warren ST.A decade of molecular studies of fragile X syndrome[J].Annu Rev Neurosci,2002,25:315-338.
[34]Deng PY,Rotman Z,Blundon JA,Cho Y,Cui J,Cavalli V,et al.FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels[J].Neuron,2013,77(4):696-711.
[35]Wang T,Bray SM,Warren ST.New perspectives on the biology of fragile X syndrome[J].Curr Opin Genet Dev,2012,22(3):256-263.
[36]Santoro MR,Bray SM,Warren ST.Molecular mechanisms of fragile X syndrome:a twenty-year perspective[J].Annu Rev Pathol,2012,7:219-245.
[37]AschrafiA, Cunningham BA, EdelmanGM,Vanderklish PW.The fragile X mental retardation protein and groupⅠmetabotropic glutamate receptors regulate levels of mRNA granules in brain[J].Proc Natl Acad Sci USA,2005,102(6):2180-2185.
[38]Osterweil EK,Krueger DD,Reinhold K,Bear MF.Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome[J].J Neurosci,2010,30(46):15616-15627.
[39]Silverman JL,Smith DG,Rizzo SJ,Karras MN,Turner SM,Tolu SS,et al.Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism[J].Sci Transl Med, 2012,4(131):131ra 51.
[40]Adusei DC,Pacey LK,Chen D,Hampson DR.Early developmental alterations in GABAergic protein expression in fragile X knockout mice[J].Neuropharmacology,2010,59(3):167-171.
[41]Wang H,Wu LJ,Kim SS,Lee FJ,Gong B,Toyoda H.FMRP acts as a key messenger for dopamine modulation in the forebrain[J].Neuron,2008,59(4):634-647.
[42]Erickson CA,Stigler KA,Wink LK,Mullett JE,Kohn A,Posey DJ,et al.A prospective open-label study of aripiprazole in fragile X syndrome[J].Psychopharmacology(Berl),2011,216(1):85-90.
[43]Lipton JO,Sahin M.The neurology of mTOR[J].Neuron,2014,84(2):275-291.
[44]Osterweil EK,Chuang SC,Chubykin AA,Sidorov M,Bianchi R,Wong RK,et al.Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome[J].Neuron,2013,77(2):243-250.
[45]Berry-Kravis E,Sumis A,Hervey C,Nelson M,Porges SW,Weng N,et al.Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome[J].J Dev Behav Pediatr,2008,29(4):293-302.
[46]Tavazoie SF,Alvarez VA,Ridenour DA,Kwiatkowski DJ,Sabatini BL.Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2[J].Nat Neurosci,2005,8(12):1727-1734.
[47]Nie D,Di Nardo A,Han JM,Baharanyi H,Kramvis I,Huynh T,et al.Tsc2-Rheb signaling regulates EphA-mediated axon guidance[J].Nat Neurosci,2010,13(2):163-172.
[48]Tang G,Gudsnuk K,Kuo SH,Cotrina ML,Rosoklija G,Sosunov A,et al.Loss ofmTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits[J].Neuron,2014,83(5):1131-1143.
[49]Crino PB.Evolving neurobiology of tuberous sclerosis complex[J].Acta Neuropathol,2013,125(3):317-332.
[50]Reith RM,McKenna J,Wu H,Hashmi SS,Cho SH,Dash PK,et al.Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex[J].Neurobiol Dis,2013,51:93-103.
[51]Tsai PT,Hull C,Chu Y,Greene-Colozzi E,Sadowski AR,Leech JM,et al.Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice[J].Nature,2012,488(7413):647-651.
[52]Ehninger D,Han S,Shilyansky C,Zhou Y,Li W,Kwiatkowski DJ,et al.Reversal of learning deficits in a Tsc2+/-mouse model of tuberous sclerosis[J].Nat Med,2008,14(8):843-848.
[53]Jones PL,Veenstra GJ,Wade PA,Vermaak D,Kass SU,Landsberger N,et al.Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription[J].Nat Genet,1998,19(2):187-191.
[54]Nan X,Ng HH,Johnson CA,Laherty CD,Turner BM,Eisenman RN,et al.Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex[J].Nature,1998,393(6683):386-389.
[55]Yasui DH,Peddada S,Bieda MC,Vallero RO,HogartA, Nagarajan RP, etal.Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes[J].Proc Natl Acad Sci USA,2007,104(49):19416-19421.
[56]Skene PJ,Illingworth RS,Webb S,Kerr AR,James KD,Turner DJ,et al.Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state[J].Mol Cell,2010,37(4):457-468.
[57]Maezawa I,Jin LW.Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate[J].J Neurosci,2010,30(15):5346-5356.
[58]Amir RE,Van den Veyver IB,Wan M,Tran CQ,Francke U,Zoghbi HY.Rett syndrome is caused by mutations in X-linked MECP2,encoding methyl-CpG-binding protein 2[J].Nat Genet,1999,23(2):185-188.
[59]Neul JL,Fang P,Barrish J,Lane J,Caeg EB,Smith EO,et al.Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome[J].Neurology,2008,70(16):1313-1321.
[60]Meins M,Lehmann J,Gerresheim F,Herchenbach J,Hagedorn M,Hameister K,et al.Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome[J].J Med Genet,2005,42(2):e12(2005-02).
[61]del Gaudio D,Fang P,Scaglia F,Ward PA,Craigen WJ,Glaze DG,et al.Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males[J].Genet Med,2006,8(12):784-792.
[62]Van Esch H,Bauters M,Ignatius J,Jansen M,Raynaud M,Hollanders K,et al.Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males[J].Am J Hum Genet,2005,77(3):442-453.
[63]Ramocki MB,Peters SU,Tavyev YJ,Zhang F,Carvalho CM,Schaaf CP,et al.Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome[J].Ann Neurol,2009,66(6):771-782.
[64]Ramocki MB,Tavyev YJ,Peters SU.The MECP2 duplication syndrome[J].Am J Med Genet A,2010,152A(5):1079-1088.
[65]Liu Z,Li X,Zhang JT,Cai YJ,Cheng TL,Cheng C,et al.Autism-like behaviours and germline transmissionintransgenicmonkeysoverexpressing MeCP2[J].Nature,2016,530(7588):98-102.
[66]Chen Y,Yu J,Niu Y,Qin D,Liu H,Li G,et al.Modeling Rett syndrome using TALEN-edited MECP2 mutant cynomolgus monkeys[J].Cell,2017,169(5):945-955.e10.
[67]Splawski I,Timothy KW,Sharpe LM,Decher N,Kumar P,Bloise R,et al.Ca(Ⅴ)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism[J].Cell,2004,119(1):19-31.
[68]Kochlamazashvili G,Henneberger C,Bukalo O,Dvoretskova E,Senkov O,Lievens PM,et al.The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca2+channels[J].Neuron,2010,67(1):116-128.
[69]Brigidi GS,Bamji SX.Cadherin-catenin adhesion complexes at the synapse[J].Curr Opin Neurobiol,2011,21(2):208-214.
[70]De Rubeis S,He X,Goldberg AP,Poultney CS,Samocha K,Cicek AE,et al.Synaptic,transcriptional and chromatin genes disrupted in autism[J].Nature,2014,515(7526):209-215.
[71]Chahrour M,Jung SY,Shaw C,Zhou X,Wong ST,Qin J,et al.MeCP2,a key contributor to neurological disease,activates and represses transcription[J].Science,2008,320(5880):1224-1229.
[72]Wu H,Tao J,Chen PJ,Shahab A,Ge W,Hart RP,et al.Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome[J].Proc Natl Acad Sci USA,2010,107(42):18161-18166.
[73]Gao Z,Lee P,Stafford JM,von Schimmelmann M,Schaefer A,Reinberg D.An AUTS2-polycomb complex activates gene expression in the CNS[J].Nature,2014,516(7531):349-354.
[74]Katayama Y,Nishiyama M,Shoji H,Ohkawa Y,Kawamura A,Sato T,et al.CHD8 haploinsufficiency results in autistic-like phenotypes in mice[J].Nature,2016,537(7622):675-679.
[75]Willsey AJ,Sanders SJ,Li M,Dong S,Tebbenkamp AT,Muhle RA,et al.Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism[J].Cell,2013,155(5):997-1007.
[76]Parikshak NN,Luo R,Zhang A,Won H,Lowe JK,Chandran V,et al.Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism[J].Cell,2013,155(5):1008-1021.
[77]Gogolla N,Leblanc JJ,Quast KB,Südhof TC,Fagiolini M,Hensch TK.Common circuit defect of excitatory-inhibitory balance in mouse models of autism[J].J Neurodev Disord,2009,1(2):172-181.
[78]Lawrence YA,Kemper TL,Bauman ML,Blatt GJ.Parvalbumin-,calbindin-,and calretinin-immunoreactive hippocampal interneuron density in autism[J].Acta Neurol Scand,2010,121(2):99-108.
[79]Rothwell PE,Fuccillo MV,Maxeiner S,Hayton SJ,Gokce O,Lim BK,et al.Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors[J].Cell,2014,158(1):198-212.
[80]Pe?a J,Feliciano C,Ting JT,Wang W,Wells MF,Venkatraman TN,et al.Shank3 mutant mice display autistic-like behaviours and striatal dysfunction[J].Nature,2011,472(7344):437-442.
[81]Asano E,Chugani DC,Muzik O,Behen M,Janisse J,Rothermel R,et al.Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction[J].Neurology,2001,57(7):1269-1277.
[82]Cheng W,Rolls ET,Gu H,Zhang J,Feng J.Autism:reduced connectivity between cortical areas involved in face expression,theory of mind,and the sense of self[J].Brain,2015,138(Pt 5):1382-1393.
[83]McPheeters ML,Warren Z,Sathe N,Bruzek JL,Krishnaswami S,Jerome RN,et al.A systematic review of medical treatments for children with autism spectrum disorders[J].Pediatrics,2011,127(5):e1312-e1321.
[84]Siegel M,Beaulieu AA.Psychotropic medications in children with autism spectrum disorders:a systematic review and synthesis for evidence-based practice[J].J Autism Dev Disord,2012,42(8):1592-1605.
[85]McCracken JT,McGough J,Shah B,Cronin P,Hong D,Aman MG,et al.Risperidone in children with autism and serious behavioral problems[J].N Engl J Med,2002,347(5):314-321.
[86]Anderson GM,Scahill L,McCracken JT,McDougle CJ,Aman MG,Tierney E,et al.Effects of shortand long-term risperidone treatment on prolactin levels in children with autism[J].Biol Psychiatry,2007,61(4):545-550.
[87]Marcus RN,Owen R,Kamen L,Manos G,McQuade RD,Carson WH,et al.A placebocontrolled,fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder[J].J Am Acad Child Adolesc Psychiatry,2009,48(11):1110-1119.
[88]Auerbach BD,Osterweil EK,Bear MF.Mutations causing syndromic autism define an axis of synaptic pathophysiology[J].Nature,2011,480(7375):63-68.
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
