一種基于位置信息的高效DNA序列挖掘算法
2017-07-10 10:27:26楊靜欣毛國(guó)君
計(jì)算機(jī)應(yīng)用與軟件 2017年6期
楊靜欣 毛國(guó)君
(中央財(cái)經(jīng)大學(xué)信息學(xué)院 北京 100081)
一種基于位置信息的高效DNA序列挖掘算法
楊靜欣 毛國(guó)君
(中央財(cái)經(jīng)大學(xué)信息學(xué)院 北京 100081)
類Apriori算法在產(chǎn)生頻繁模式時(shí)需要多次掃描數(shù)據(jù)庫,并且產(chǎn)生大量的候選集;FreeSpan和PrefixSpan等基于投影數(shù)據(jù)庫的算法在產(chǎn)生頻繁模式時(shí)會(huì)產(chǎn)生大量的投影數(shù)據(jù)庫,占用很多內(nèi)存空間,這些都造成了很大的冗余。針對(duì)以往序列挖掘算法存在的不足,提出一種高效的序列挖掘算法——基于位置信息的序列挖掘算法PBSMA(Position-Based Sequence Mining Algorithm)。PBSMA算法通過記錄頻繁子序列的位置信息來減少對(duì)數(shù)據(jù)庫的掃描,利用位置信息逐漸擴(kuò)大頻繁模式的長(zhǎng)度,并且借鑒關(guān)聯(lián)矩陣的思想和PrefixSpan算法中前綴的概念,深度優(yōu)先去尋找更長(zhǎng)的關(guān)鍵模式。實(shí)驗(yàn)結(jié)果證明,無論在時(shí)間還是空間上,PBSMA算法都比PrefixSpan算法更高效。
序列挖掘 DNA序列 位置信息 關(guān)聯(lián)矩陣 前綴
0 引 言
序列挖掘是數(shù)據(jù)挖掘中非常重要的一個(gè)研究領(lǐng)域,一般是指從序列數(shù)據(jù)庫中發(fā)現(xiàn)蘊(yùn)含在其中的,相對(duì)于時(shí)間或者其他順序高頻率出現(xiàn)的子序列,并稱這個(gè)高頻子序列為序列模式。序列挖掘的概念最早由Agrawal等在針對(duì)超市中購(gòu)物籃數(shù)據(jù)的分析中提出[1],目的是發(fā)現(xiàn)某一時(shí)間段內(nèi)客戶的購(gòu)買活動(dòng)規(guī)律。序列挖掘經(jīng)過近20年的發(fā)展,其應(yīng)用范圍已經(jīng)不再局限于交易數(shù)據(jù)庫,在商業(yè),消費(fèi)者行為分析,股票趨勢(shì)預(yù)測(cè),生物序列分析,Web挖掘等領(lǐng)域都有重要的應(yīng)用。商業(yè)組織利用序列模式去研究客戶購(gòu)買行為模式特征、計(jì)算生物學(xué)中序列模式挖掘來分析不同氨基酸突變模式、用戶Web訪問模式預(yù)測(cè)以及DNA序列分析和譜分析,尤其在DNA分析等尖端科學(xué)研究領(lǐng)域、Web訪問等新型應(yīng)用數(shù)據(jù)源等方面得到了有針對(duì)性的研究[2-3]。
DNA序列挖掘是序列挖掘最重要的應(yīng)用之一,通過挖掘DNA序列,研究者可以發(fā)現(xiàn)隱藏在序列數(shù)據(jù)背后的有價(jià)值的規(guī)律,用以解讀相應(yīng)生物體中的生命特征。眾所周知,生物的遺傳信息被儲(chǔ)存在DNA中,而DNA是由兩條盤繞在雙螺旋結(jié)構(gòu)上的線性鏈組成,每條鏈可以看成是四種核苷酸A(腺嘌呤)、G(鳥嘌呤)、T(胸腺嘧啶)、C(胞嘧啶)的線性組合,兩條線性鏈嚴(yán)格遵守堿基配對(duì)規(guī)則,即A與T配對(duì)、C與G配對(duì)出現(xiàn)。例如,一條DNA序列是
二十一世紀(jì)是生物技術(shù)的時(shí)代,生物技術(shù)是當(dāng)前最熱門的研究領(lǐng)域之一[4]。DNA數(shù)據(jù)量龐大,其中蘊(yùn)藏著海量的信息,我們有理由相信,DNA序列挖掘?qū)?huì)幫助人類解開更多的基因之謎,在遺傳、疾病、物種學(xué)上帶來更多的突破。DNA全序列具有什么樣的結(jié)構(gòu)、由這4個(gè)字符排成的看似隨機(jī)的序列中隱藏著什么規(guī)律、各堿基的功能等都有待于進(jìn)一步的研究。因此,DNA序列模式挖掘成為序列挖掘最重要、最有價(jià)值的應(yīng)用。
1 相關(guān)工作
在早期,序列挖掘算法大多基于Agrawal等提出的關(guān)聯(lián)規(guī)則算法—Apriori算法[5],該算法的核心理論是頻繁模式的任何子模式都是頻繁的。Agrawal和Srikant提出的AprioriAll算法、AprioriSome算法和DynamicSome算法都是基于這個(gè)思想而提出,成為序列挖掘算法研究的基礎(chǔ)[1];1996年,Srikant和Agrawal又提出GSP算法,這是一種廣度優(yōu)先的自下而上的算法,采用不同的剪枝策略產(chǎn)生較少的候選模式,因而效率相對(duì)于AprioriAll算法有所提高,是目前引用率較高的算法之一[6];然而類Apriori算法自身存在局限性,學(xué)者們紛紛開始尋找更高效的方法。2000年,Han等提出了基于模式增長(zhǎng)的FP-growth算法,該算法不產(chǎn)生候選集,而是使用FP-tree的樹狀結(jié)構(gòu)壓縮數(shù)據(jù)庫,然后再利用FP-tree對(duì)頻繁模式從下向上的挖掘,加快了挖掘過程[7]。隨后,學(xué)者們又提出了一系列基于數(shù)據(jù)投影的算法,包括Han和Pei于2000年提出的FreeSpan算法[8]和2001年提出的PrefixSpan算法[9],這些都是經(jīng)典的序列挖掘算法。近年來,在經(jīng)典序列挖掘算法的基礎(chǔ)上,學(xué)者們相繼提出了優(yōu)化的序列挖掘算法。2007年,張坤等提出一種基于GSP的MFS(Mining Frequent Sequence)算法[10],它不需要多次遍歷數(shù)據(jù)庫,通過連接不同長(zhǎng)度的已知頻繁序列來產(chǎn)生的候選序列,比GSP算法更高效;張利軍等提出一種基于位置信息的序列模式挖掘算法PVS,在使用PrefixSpan算法時(shí)通過記錄每個(gè)已產(chǎn)生的投影數(shù)據(jù)庫的位置信息降低投影數(shù)據(jù)庫的冗余,提高算法效率[11];2012年,劉棟等提出了基于Map Reduce的序列挖掘模式,在PrefixSpan算法的基礎(chǔ)上,對(duì)模式挖掘任務(wù)進(jìn)行分割,利用Map函數(shù)處理由不同前綴得到的序列模式并構(gòu)造投影數(shù)據(jù)庫,從而提高挖掘效率及簡(jiǎn)化搜索空間[12];2013年,吳信東等設(shè)計(jì)了一種帶有通配符的模式挖掘算法One-Off Mining,通配符的加入使模式的形式更加靈活[13];2015年,劉端陽等首次在頻繁序列模式挖掘算法中引入邏輯的思想,提出一種基于邏輯的頻繁序列挖掘算法LFSPM,通過邏輯規(guī)則過濾,優(yōu)化結(jié)果集[14]。
DNA序列挖掘作為序列挖掘領(lǐng)域最重要的應(yīng)用之一,也獲得了很多學(xué)者的特別關(guān)注。學(xué)者們結(jié)合DNA序列的特點(diǎn)后對(duì)傳統(tǒng)的序列挖掘算法進(jìn)行了有意識(shí)的改造提出了在處理DNA序列是更高效的算法。2007年,朱揚(yáng)勇等對(duì)于DNA序列挖掘的問題進(jìn)行了綜述,回顧了DNA序列挖掘的發(fā)展歷程,總結(jié)出DNA序列挖掘經(jīng)歷了三個(gè)階段,依次是基于統(tǒng)計(jì)技術(shù)的應(yīng)用階段,一般化挖掘方法的應(yīng)用階段到如今的專門面向DNA序列挖掘的設(shè)計(jì)階段[15];2007年,熊赟等提出了一種融合自底向上和自頂向下策略挖掘DNA重復(fù)序列的新算法DnaReSM[16],克服了完全自底向上算法產(chǎn)生大量短候選模式的缺點(diǎn);2011年,周溜溜等提出了一種基于頻繁子樹挖掘策略的DNA頻繁模式挖掘算法,繞開了傳統(tǒng)序列的對(duì)比方式,將序列按照后綴樹結(jié)構(gòu)方式進(jìn)行組織約減提升識(shí)別效率[17];2013年,姜華等在引入近似度的概念的基礎(chǔ)上,構(gòu)造頻繁近似模式,提出了頻繁近似模式的挖掘算法SFAP[18];2015年,毛國(guó)君等在分析DNA序列的特點(diǎn)的基礎(chǔ)上,提出了一種基于關(guān)聯(lián)矩陣(AMSMA)的算法,將掃描數(shù)據(jù)庫得到的DNA序列信息儲(chǔ)存在關(guān)聯(lián)矩陣(Association Marix)中,利用矩陣壓縮空間使用率,體現(xiàn)出較好的時(shí)間和空間效率[19]。這些算法關(guān)注于DNA序列的特點(diǎn),大大提升了算法的效率。
本文中汲取了上面多個(gè)算法的精華,分別是PrefixSpan算法中基于前綴的思想[9]、PVS算法中位置信息的概念[11]和AMSMA算法中關(guān)聯(lián)矩陣的概念[19],利用這些算法的優(yōu)勢(shì)提出一種高效的基于位置信息的DNA序列挖掘算法PBSMA(Position-Based Sequence Mining Algorithm)。
2 PBSMA算法描述
2.1 基本概念
定義1 DNA序列(DNA Sequence):給定字符集合E={A,T,C,G},一個(gè)DNA序列被表示為S=
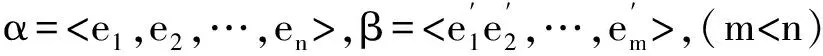
例如,在序列abacd中,aba是abacd的前綴,而ba不是。
定義3 關(guān)聯(lián)矩陣:給定DNA序列S=
例如在序列s=
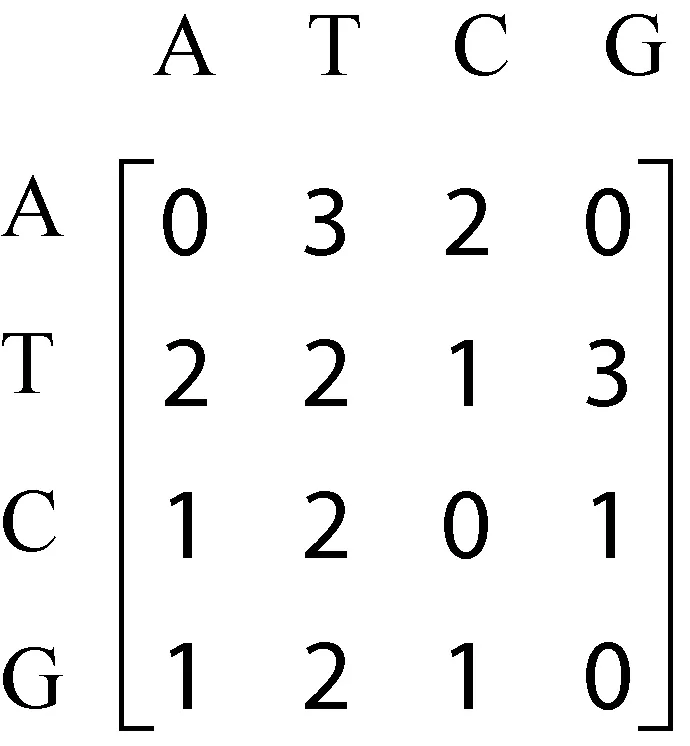
圖1 關(guān)聯(lián)矩陣舉例
定義4 位置信息:一個(gè)子序列在數(shù)據(jù)庫S中的位置信息為這個(gè)子序列在數(shù)據(jù)庫S中的序列號(hào)和在序列中的結(jié)束位置,記為(s,v),其中s表示S中包含該模式序列的序號(hào);v表示對(duì)應(yīng)的序列關(guān)于該模式的結(jié)束位置。當(dāng)一個(gè)子序列在數(shù)據(jù)庫中多次出現(xiàn)時(shí),這個(gè)子序列的位置信息表示為((s1,v1),(s2,v2),…,(sn,vn))。
2.2 算法思路
PBSMA算法是一種基于前綴的深度優(yōu)先的挖掘算法,以DNA數(shù)據(jù)為例,在使用PBSMA算法處理上,首先通過掃描數(shù)據(jù)庫來獲取大1-項(xiàng)集L1={A,T,C,G},然后依次以L1中的元素為前綴構(gòu)造關(guān)聯(lián)矩陣,例如先以A為前綴產(chǎn)生所有以A開頭的關(guān)鍵模式,深度優(yōu)先直到不再有關(guān)鍵模式產(chǎn)生時(shí),再以T為前綴……依次類推,直到遍歷過L1中的所有元素后停止。
PBSMA算法的步驟如下:
(1) 掃描序列數(shù)據(jù)庫,得到長(zhǎng)度為1的序列模式L1,作為初始的種子集;
(2) 對(duì)于L1中的每一個(gè)元素,依次取出一個(gè)元素作為新候選模式的前綴,以此來劃分搜索空間;
(3) 將上一步得到的前綴作為關(guān)聯(lián)矩陣的行,以L1中的所有元素作為關(guān)聯(lián)矩陣的列,生成關(guān)聯(lián)矩陣。計(jì)算關(guān)聯(lián)矩陣中每一個(gè)候選模式在數(shù)據(jù)庫中出現(xiàn)的次數(shù)(即支持度)并保存每次出現(xiàn)的位置信息;
(4) 檢測(cè)關(guān)聯(lián)矩陣中候選模式的支持度是否大于設(shè)定的最小支持度,若大于,則該子序列為關(guān)鍵序列,加入到L2并更新其位置信息;若不大于,則不是關(guān)鍵序列,刪除之前保存的位置信息;
(5) 利用L2和位置信息依次生成更高階關(guān)聯(lián)矩陣,得到L3,L4……直到?jīng)]有新的序列模式或新的候選序列模式產(chǎn)生為止。
重復(fù)步驟(2)-步驟(5),直到遍歷完L1中的所有序列。
值得注意的是,與從單條序列中挖掘關(guān)鍵模式不同,在多條序列的挖掘中,如果某子序列在一條記錄中多次出現(xiàn),仍然只計(jì)數(shù)一次,但需要記錄該子序列出現(xiàn)的每一個(gè)位置信息,后面的例子中將會(huì)詳細(xì)說明這一點(diǎn)。
2.3 算法舉例
例1 例如在表1中的DNA序列數(shù)據(jù)庫,設(shè)最小支持度為2,則子序列AT是一個(gè)頻繁模式,它的位置記為((1,2)、(1,7)、(2,4)、(2,9)、(3,4))。其中在(1,2)中,1表示S中的第一條序列,2中表示AT這個(gè)子序列的最后一個(gè)字符在序列1中所處的位置為2,后四者亦同。

表1 例1中DNA序列數(shù)據(jù)庫
在上例中,首先構(gòu)造A為前綴的關(guān)聯(lián)矩陣,構(gòu)造關(guān)聯(lián)矩陣如圖2所示。
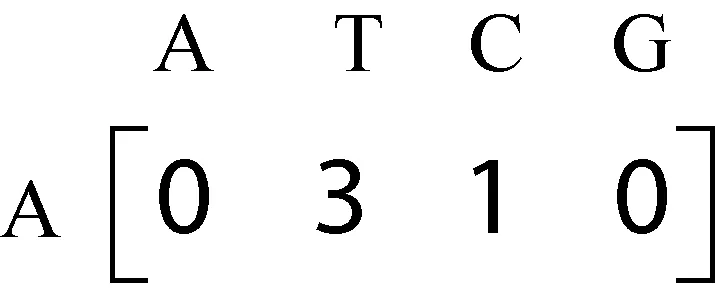
圖2 前綴為A的一階關(guān)聯(lián)矩陣
掃描計(jì)數(shù)的同時(shí),記錄到的AT的位置信息(1,2)、(1,7)、(2,4)、(2,8)和(3,4),AC的位置信息(3,7)。當(dāng)支持度為2時(shí),AT是頻繁模式,而AC不是,因此進(jìn)行剪枝,不再保留AC的位置信息。
利用上一步得到的長(zhǎng)度為2的頻繁模式構(gòu)造2階關(guān)聯(lián)矩陣。此時(shí)無需掃描數(shù)據(jù)庫,利用上一步得到的位置信息,直接去序列中尋找AT的后綴,構(gòu)造關(guān)聯(lián)矩陣并計(jì)數(shù),同時(shí)更新位置信息。在上例中,出現(xiàn)在(1,2)位置的AT直接后綴為T,則在關(guān)聯(lián)矩陣相應(yīng)位置將子序列ATT的支持度增加1,并保存位置信息(1,3),出現(xiàn)在(2,4)位置的AT直接后綴為C,則子序列ATC的支持度增加1,并保存位置信息(2,5)…依次找到每一個(gè)位置信息下的AT的后綴形成關(guān)聯(lián)矩陣并保存位置信息,得到如圖3的二階關(guān)聯(lián)矩陣。
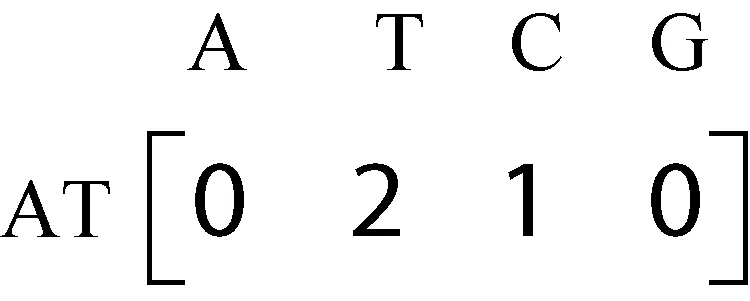
圖3 以A為前綴的二階關(guān)聯(lián)矩陣
對(duì)應(yīng)位置信息為ATT(1,3)、(1,8)、(3,5),ATC(2,5)。在支持度為2時(shí),得到長(zhǎng)度為3的頻繁序列ATT,刪除ATC的位置信息。同樣,利用長(zhǎng)度為3的頻繁序列構(gòu)造4階關(guān)聯(lián)矩陣,利用位置信息找到長(zhǎng)度為4的候選序列,計(jì)數(shù)并記錄位置信息后發(fā)現(xiàn)長(zhǎng)度為4的頻繁模式ATTA,位置信息為(1,9)、(2,6),如圖4所示。利用長(zhǎng)度為4的頻繁模式構(gòu)造五階關(guān)聯(lián)矩陣,如圖5所示,沒有發(fā)現(xiàn)支持度大于2的頻繁模式。至此,以A為前綴的頻繁模式全部找到。
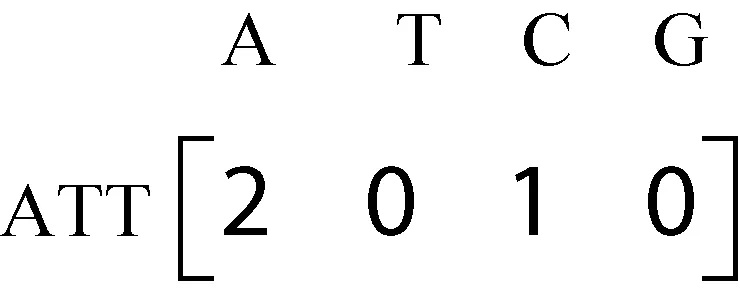
圖4 以A為前綴的三階關(guān)聯(lián)矩陣
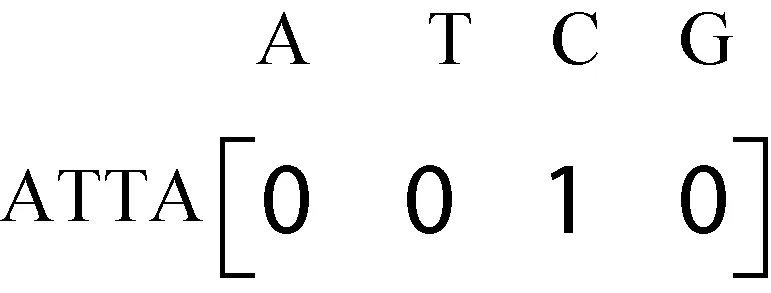
圖5 以A為前綴的四階關(guān)聯(lián)矩陣
同樣的,應(yīng)用上面的方法分別建立以T、C、G為前綴的各階關(guān)聯(lián)矩陣,采用深度優(yōu)先的原則分別發(fā)現(xiàn)以T、C、G為前綴的頻繁模式。產(chǎn)生的所有頻繁模式如表2所示。

表2 例1中的所有頻繁模式
由此可得到最大關(guān)鍵模式有:CTA,TCG,CGATTA。
2.4 算法偽代碼
算法描述:PBSMA算法偽碼
輸入:稀疏項(xiàng)目集序列數(shù)據(jù)庫S;最小支持度min-sup
輸出:S的關(guān)鍵子序列KS
1. scan S to get L1{};
//掃描數(shù)據(jù)庫找到所有的頻繁1-序列
2. FOR m=0 TO L1.size()
//深度優(yōu)先,最外層循環(huán)依次遍歷L1中的每個(gè)序列
3. former=L1.get(m);
4. FOR n=0 TO L1.size();
//該層循環(huán)意在獲取初始位置
5. later=L1.get(n);
6. string=former+later;
//子序列連接
7. Calculate support of string occur in S ;
//計(jì)算子序列支持度
8. IF(support >=min_sup)
//大于等于最小支持度為頻繁模式
9. add string to KS;
10. save Position of string;
//記錄子序列位置信息
11. WHEN(Position is not empty) DO
12. use Position to extend longer subsequence;
13. calculate support;
//利用位置信息尋找更長(zhǎng)的頻繁模式并計(jì)算支持度
14. IF support>=min-sup
15. add subsequence to KS;
16. update Positon;
//更新位置信息
17. ELSE
18. delete Position;
//不滿足最小支持度的序列刪除位置信息
19. END DO
20. Retrun KS
//輸出所有頻繁序列
3 實(shí)驗(yàn)分析
本部分實(shí)驗(yàn)中DNA數(shù)據(jù)來源于美國(guó)國(guó)家生物技術(shù)信息中心(http://www.ncbi.nim.nih.gov)。由于這里只探討算法的可用性和時(shí)間空間效率,而不是要解決生物問題,也無需對(duì)實(shí)驗(yàn)結(jié)果進(jìn)行解釋,因此為了更好地控制實(shí)驗(yàn)變量,實(shí)驗(yàn)數(shù)據(jù)在原始數(shù)據(jù)上進(jìn)行了一定的處理,具體處理方法在下面的各個(gè)實(shí)驗(yàn)中作了說明。
實(shí)驗(yàn)是在一臺(tái)4GB內(nèi)存、使用英特爾酷睿i3,1.40 GHz處理器的計(jì)算機(jī)上進(jìn)行。采用的對(duì)比算法是同樣基于分治思想的PrefixSpan算法[9]。實(shí)驗(yàn)的目標(biāo)主要是檢測(cè)本文算法的精度和效率(包括時(shí)間效率和空間效率)。
實(shí)驗(yàn)1 比較內(nèi)存空間使用情況
由于PBSMA算法不需要產(chǎn)生投影數(shù)據(jù)庫,因此占用更少的內(nèi)存。實(shí)驗(yàn)1模擬PreifixSpan算法和PBSMA算法的運(yùn)行過程,對(duì)實(shí)驗(yàn)1中的序列數(shù)據(jù)進(jìn)行分析,每一步的占用內(nèi)存如表3所示。由于兩個(gè)算法都是基于前綴的分治思想,因此表3中先討論以L1中頻繁1-序列為前綴的情況,最后再?gòu)目傮w上進(jìn)行討論。
值得注意的是,PrefixSpan算法的投影數(shù)據(jù)庫保存投影序列,在本實(shí)驗(yàn)中即字符序列,是字符型數(shù)據(jù);PBSMA算法的位置信息保存坐標(biāo)位置,由兩個(gè)整型數(shù)值構(gòu)成,為方便表示,在表3的討論中均將其作為字符處理。

表3 PrefixSpan算法和PBSMA算法的內(nèi)存使用情況
從表3的實(shí)驗(yàn)結(jié)果可以看出,無論利用分治的思想在每一個(gè)小范圍內(nèi)討論,還是從總體上考慮最大使用內(nèi)存和合計(jì)使用內(nèi)存情況,PBSMA算法的空間效率都要好于PrefixSpan算法。雖然PBSMA算法需要記錄較多的位置信息,但由于每個(gè)位置信息僅占用兩字符的內(nèi)存,相比于PrefixSpan算法投影數(shù)據(jù)庫中的子序列相比,仍然有優(yōu)勢(shì)。
實(shí)驗(yàn)1中序列數(shù)據(jù)量小,并且每條序列長(zhǎng)度較短,當(dāng)應(yīng)用于實(shí)際的序列數(shù)據(jù)庫時(shí),PrefixSpan算法將產(chǎn)生更多的投影數(shù)據(jù)庫,因而PBSMA算法的空間優(yōu)勢(shì)將會(huì)更明顯。
實(shí)驗(yàn)2 比較不同的最小支持度下執(zhí)行時(shí)間
本實(shí)驗(yàn)的數(shù)據(jù)來源于美國(guó)國(guó)家生物技術(shù)信息中心(NCBI)的序列數(shù)據(jù)庫,序列被隨機(jī)劃分為長(zhǎng)度20~30的序列,實(shí)驗(yàn)記錄1 369條,共28 739個(gè)字符,將此實(shí)驗(yàn)數(shù)據(jù)集命名為PBSMA實(shí)驗(yàn)數(shù)據(jù)集-1。
利用PBSMA實(shí)驗(yàn)數(shù)據(jù)集-1,在兩個(gè)最小支持度區(qū)間進(jìn)行實(shí)驗(yàn),分別是小支持度區(qū)間(5%~30%)、大支持度區(qū)間(10%~90%),測(cè)試本文提出的PBSMA算法和PrefixSpan算法的運(yùn)行效率。表4給出的是實(shí)驗(yàn)數(shù)據(jù)在小支持度區(qū)間范圍內(nèi)挖掘出的頻繁序列的個(gè)數(shù),圖6對(duì)應(yīng)給出在該區(qū)間內(nèi)PrefixSpan和PBSMA運(yùn)行時(shí)間效率的對(duì)比;表5給出的是實(shí)驗(yàn)數(shù)據(jù)在大支持度區(qū)間范圍內(nèi)挖掘出的頻繁序列的個(gè)數(shù),圖7對(duì)應(yīng)給出在大支持度區(qū)間PrefixSpan和PBSMA運(yùn)行時(shí)間效率的對(duì)比。
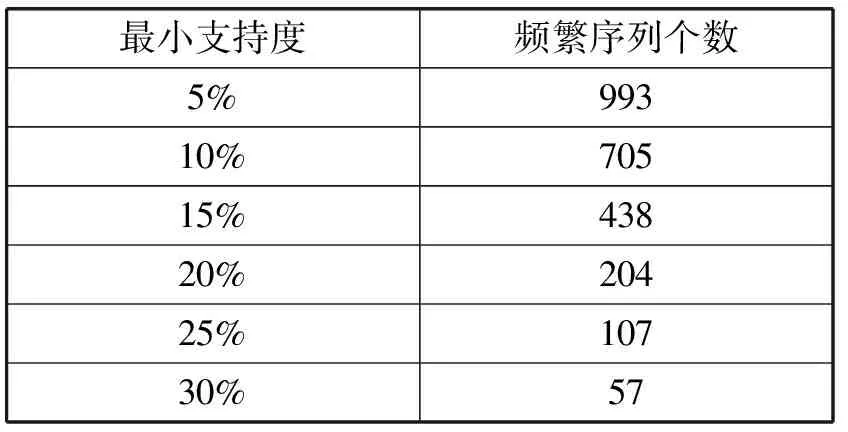
表4 小支持度區(qū)間內(nèi)挖掘出頻繁序列個(gè)數(shù)

圖6 小支持度區(qū)間內(nèi)算法時(shí)間效率對(duì)比
由表4和圖6可以看出,在小支持度區(qū)間,實(shí)驗(yàn)數(shù)據(jù)集-1產(chǎn)生較多的關(guān)鍵模式。隨著最小支持度的增加,PBSMA和PrefixSpan算法的執(zhí)行時(shí)間都在下降,這是因?yàn)殡S最小支持度的增加,生成的關(guān)鍵子序列在減少,并且在該數(shù)據(jù)集中,關(guān)鍵子序列減少的速度很快。同時(shí)也可以看出,PBSMA算法在處理DNA序列時(shí)時(shí)間效率明顯高于PrefixSpan算法。
表5和圖7從更宏觀、更全面的角度進(jìn)行實(shí)驗(yàn),展現(xiàn)了最小支持度在更大范圍內(nèi)變化時(shí)兩算法的執(zhí)行效率對(duì)比。實(shí)驗(yàn)結(jié)果表明:隨著最小支持度的增加,PBSMA和PrefixSpan算法的執(zhí)行時(shí)間都在下降,同時(shí),PBSMA算法在各支持度下的時(shí)間效率明顯都要高于PrefixSpan算法。其中,在支持度在10%~30%的范圍內(nèi)變化時(shí),由于挖掘出的頻繁子序列數(shù)量減少速度很快,因此算法執(zhí)行時(shí)間下降速度很快;在40%~70%的范圍內(nèi),頻繁子序列數(shù)量減少速度相對(duì)較慢,因此挖掘算法的執(zhí)行時(shí)間降速放緩甚至趨于平穩(wěn);然而在70%及以上的支持度下,由于此時(shí)已經(jīng)不再有頻繁模式被挖掘出來,算法執(zhí)行時(shí)間再一次驟減,并且由于兩算法在掃描一次數(shù)據(jù)庫后基本不用再進(jìn)行后面的循環(huán),因此兩算法的運(yùn)行時(shí)間都趨于0,PBSMA算法的優(yōu)勢(shì)也不如產(chǎn)生大量頻繁子序列時(shí)明顯。
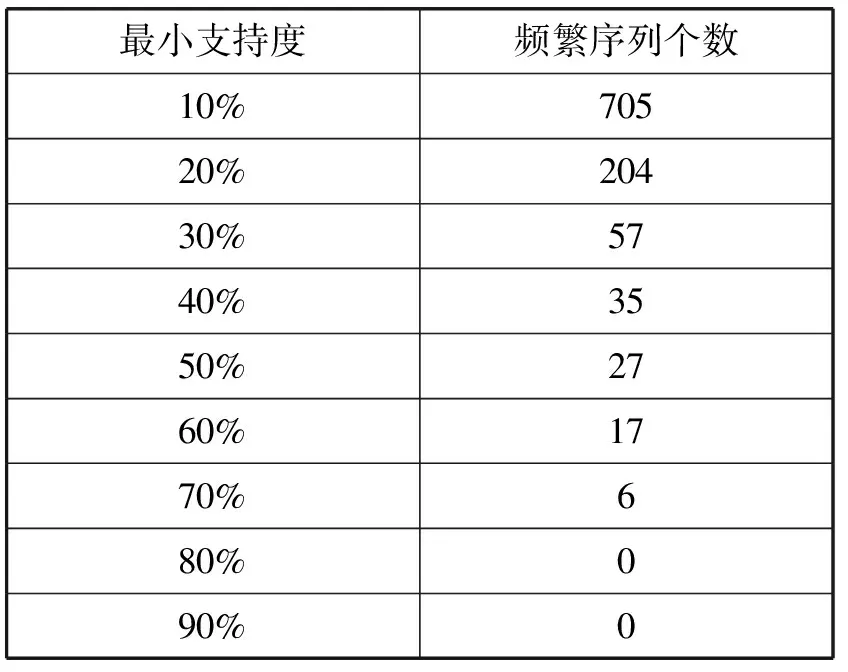
表5 大支持度區(qū)間內(nèi)挖掘出頻繁序列個(gè)數(shù)
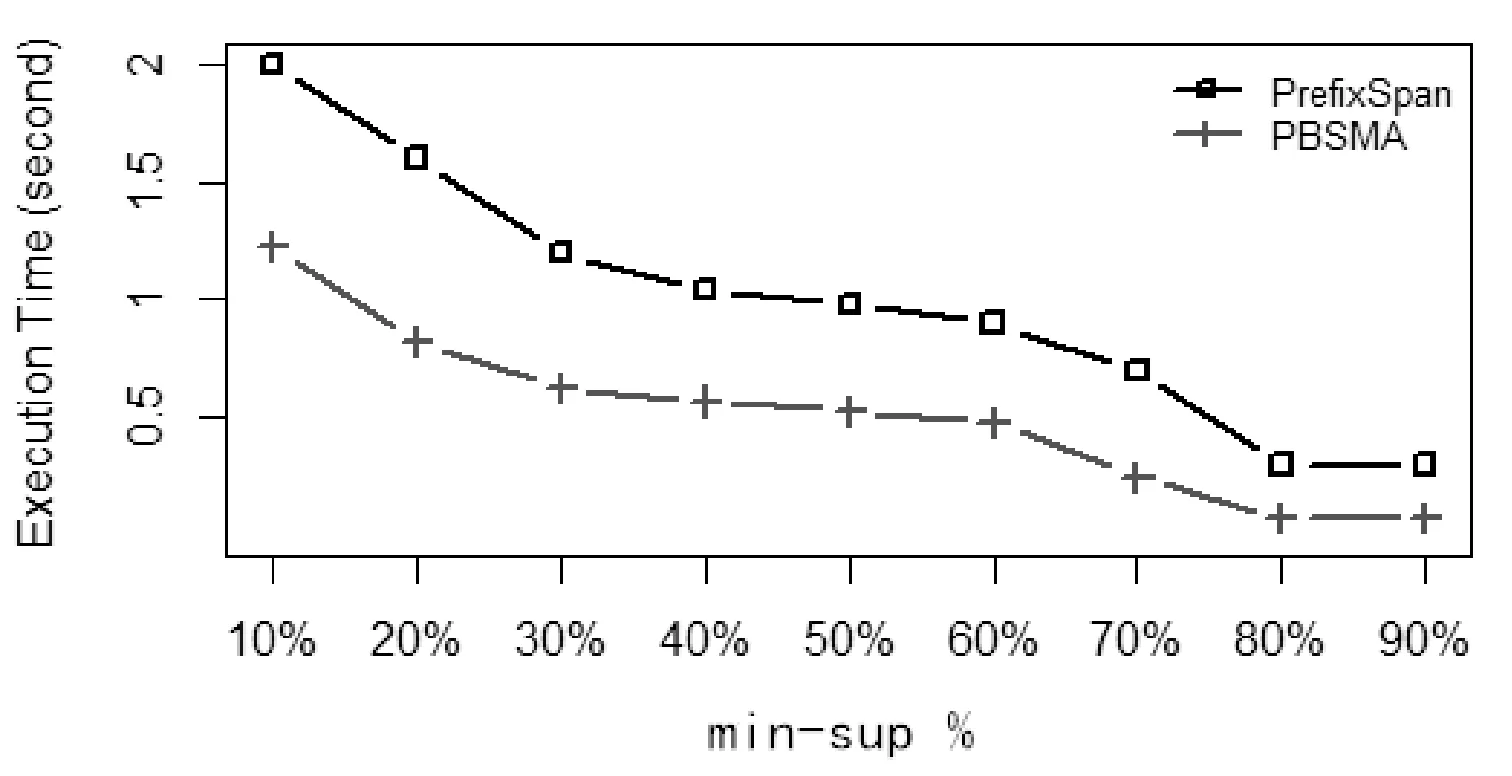
圖7 大支持度區(qū)間內(nèi)算法時(shí)間效率對(duì)比
實(shí)驗(yàn)3 比較不同的DNA序列數(shù)下執(zhí)行時(shí)間
本實(shí)驗(yàn)的數(shù)據(jù)來源于美國(guó)國(guó)家生物技術(shù)信息中心(NCBI)的序列數(shù)據(jù)庫,創(chuàng)建5個(gè)數(shù)據(jù)文件,其中存放序列數(shù)量分別為2 000、4 000、6 000、8 000、10 000,每條序列長(zhǎng)度20~30。將此實(shí)驗(yàn)數(shù)據(jù)集命名為PBSMA實(shí)驗(yàn)數(shù)據(jù)集-2。
本實(shí)驗(yàn)在控制最小支持度變量為20%不變的前提下,運(yùn)用PBSMA實(shí)驗(yàn)數(shù)據(jù)集-2中五個(gè)不等長(zhǎng)數(shù)據(jù)文件,考察隨著序列數(shù)量攀升的情況下兩算法的執(zhí)行效率。實(shí)驗(yàn)結(jié)果如圖8所示。
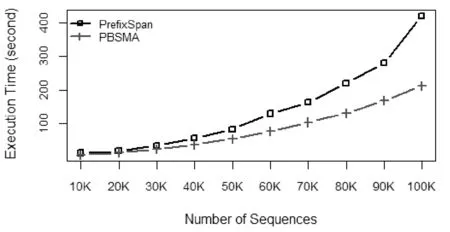
圖8 序列數(shù)量增加時(shí)執(zhí)行時(shí)間的比較
圖8表明:在固定的最小支持度下,隨著序列數(shù)據(jù)庫規(guī)模的增大,即序列數(shù)量的增加,PBSMA和PrefixSpan算法的執(zhí)行時(shí)間在攀升。在數(shù)據(jù)量較小的情況下,兩算法的運(yùn)行時(shí)間效率相差不多,但在數(shù)據(jù)量逐漸增多的過程中,PBSMA展現(xiàn)出越來越優(yōu)于PBSMA算法的表現(xiàn),特別地,當(dāng)序列數(shù)量為100 K時(shí),PrefixSpan算法所需要的時(shí)間幾乎是PBSMA算法運(yùn)行時(shí)間的兩倍,并且可以預(yù)見的是,當(dāng)數(shù)據(jù)量更大時(shí),PBSMA算法的優(yōu)勢(shì)將更加明顯。因此可以得出結(jié)論,在任何數(shù)據(jù)量下,PBSMA的算法運(yùn)行效率都要優(yōu)于PrefixSpan算法,并且數(shù)據(jù)量越大,PBSMA算法在時(shí)間效率上的提升越明顯。
4 結(jié) 語
本文提出了基于位置信息的序列挖掘算法——PBSMA算法。PBSMA算法結(jié)合了關(guān)聯(lián)矩陣、基于前綴、位置信息等的思想,用于從多條序列中挖掘出頻繁模式。較之PrefixSpan算法,PBSMA算法有如下的優(yōu)點(diǎn):
(1) 不需要產(chǎn)生投影數(shù)據(jù)庫,大大節(jié)約了存儲(chǔ)空間;
(2) 由位置信息直接定位到序列數(shù)據(jù)庫中,不需要多次掃描數(shù)據(jù)庫及投影數(shù)據(jù)庫,減少時(shí)間花銷。
在實(shí)驗(yàn)中,PBSAM算法展現(xiàn)出高于PrefixSpan算法的時(shí)間和空間效率,證明了算法的有效性和高效性。但是,本文提出的序列挖掘算法在時(shí)間和空間上都還有提升的空間,算法也可以尋找更廣闊的應(yīng)用空間,這都是未來應(yīng)該著重研究的領(lǐng)域。
[1] Agrawal R,Srikant R.Mining sequential patterns[C]//Proceedings of the 11th International Conference on Data Engineering.IEEE Computer Society,1995:3-14.
[2] 毛國(guó)君,段麗娟,王實(shí),等.數(shù)據(jù)挖掘原理與算法[M].2版.北京:清華大學(xué)出版社,2007:217-218.
[3] 陳卓,楊炳儒,宋威,等.序列模式挖掘綜述[J].計(jì)算機(jī)應(yīng)用研究,2008,25(7):1960-1963,1976.
[4] 羅春雨,毛國(guó)君,邱洪君.序列分析技術(shù)在DNA序列挖掘中的應(yīng)用[J].計(jì)算機(jī)系統(tǒng)應(yīng)用,2005(12):22-25.
[5] Agrawal R,Imieliński T,Swami A.Mining association rules between sets of items in large databases[N].ACM SIGMOD Record,1993,22(2):207-216.
[6] Srikant R,Agrawal R.Mining sequential patterns:generalizations and performance improvements[C]//Proceedings of the 5th International Conference on Extending Database Technology:Advances in Database Technology,1996:3-17.
[7]HanJ,PeiJ,YinY.Miningfrequentpatternswithoutcandidategeneration[C]//Proceedingsofthe2000ACMSIGMODInternationalConferenceonManagementofData.NewYork,NY,USA:ACMPress,2000:1-12.
[8]HanJ,PeiJ,Mortazavi-AslB,etal.FreeSpan:frequentpattern-projectedsequentialpatternmining[C]//Proceedingsofthe6thACMSIGKDDInternationalConferenceonKnowledgeDiscoveryandDataMining.NewYork,NY,USA:ACMPress,2000:355-359.
[9]PeiJ,HanJ,Mortazavi-AslB,etal.PrefixSpan:miningsequentialpatternsefficientlybyprefix-projectedpatterngrowth[C]//Proceedingsofthe17thInternationalConferenceonDataEngineering.IEEEComputerSociety,2001:215-224.
[10] 張坤,朱揚(yáng)勇.無重復(fù)投影數(shù)據(jù)庫掃描的序列模式挖掘算法[J].計(jì)算機(jī)研究與發(fā)展,2007,44(1):126-132.
[11] 張利軍,李戰(zhàn)懷,王淼.基于位置信息的序列模式挖掘算法[J].計(jì)算機(jī)應(yīng)用研究,2009,26(2):529-531.
[12] 劉棟,尉永清,薛文娟.基于MapReduce的序列模式挖掘算法[J].計(jì)算機(jī)工程,2012,38(15):43-45.
[13] 吳信東,謝飛,黃詠明,等.帶通配符和One-Off條件的序列模式挖掘[J].軟件學(xué)報(bào),2013,24(8):1804-1815.
[14] 劉端陽,馮建,李曉粉.一種基于邏輯的頻繁序列模式挖掘算法[J].計(jì)算機(jī)科學(xué),2015,42(5):260-264.
[15] 朱揚(yáng)勇,熊赟.DNA序列數(shù)據(jù)挖掘技術(shù)[J].軟件學(xué)報(bào),2007,18(11):2766-2781.
[16] 熊赟,陳越,朱揚(yáng)勇.DnaReSM:一個(gè)基于多支持度的DNA重復(fù)序列挖掘算法[J].計(jì)算機(jī)科學(xué),2007,34(2):211-212,封四.
[17] 周溜溜,業(yè)寧,徐昇,等.基于頻繁子樹挖掘的DNA重復(fù)序列識(shí)別方法[J].微電子學(xué)與計(jì)算機(jī),2011,28(9):193-196,201.
[18] 姜華,孟志青,周克江.DNA序列頻繁近似模式挖掘[J].生物信息學(xué),2013,11(1):11-15.
[19] 毛國(guó)君,楊靜欣.一種基于關(guān)聯(lián)矩陣的高效DNA序列挖掘算法[J].計(jì)算機(jī)科學(xué)與應(yīng)用,2015,5(8):271-277.
AN EFFICIENT POSITION-BASED DNA SEQUENCE MINING ALGORITHM
Yang Jingxin Mao Guojun
(SchoolofInformation,CentralUniversityofFinanceandEconomics,Beijing100081,China)
Similar to Apriori algorithm in generating frequent patterns need to scan the database several times, and generate a large number of candidate sets. Algorithms based on the projection database, such as FreeSpan and PrefixSpan, in generating frequent patterns will produce a large number of projection database, taking up a lot of memory space, which have caused a lot of redundancy. Aiming at the shortcomings of the previous sequence mining algorithms, an efficient sequence mining algorithm named PBSMA is proposed in this paper. The PBSMA reduces the scanning of the database by recording the position information of frequent subsequences, and gradually enlarges the length of the frequent patterns by using the position information. The algorithm uses the idea of association matrix and the concept of prefix in PrefixSpan algorithm to search for a longer key pattern. The experimental results show that the PBSMA is more efficient than PrefixSpan algorithm both in time and space.
Sequence mining DNA sequence Position information Association matrix Prefix
2016-05-17。國(guó)家自然科學(xué)基金項(xiàng)目(61273293)。楊靜欣,碩士生,主研領(lǐng)域:數(shù)據(jù)挖掘和生物信息學(xué)。毛國(guó)君,教授。
TP311.1
A
10.3969/j.issn.1000-386x.2017.06.042
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
甘肅教育(2020年14期)2020-09-11 07:57:42
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
財(cái)經(jīng)(2017年2期)2017-03-10 14:35:35
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
財(cái)經(jīng)(2016年15期)2016-06-03 07:38:02
財(cái)經(jīng)(2016年3期)2016-03-07 07:44:46
財(cái)經(jīng)(2016年6期)2016-02-24 07:41:51
時(shí)代英語·高二(2015年1期)2015-03-16 00:08:11
