散發型克雅病7例患者的臨床、腦電圖及影像學分析
2017-08-07 05:23:53郭方亮胡社靜李濤
卒中與神經疾病 2017年3期
郭方亮 胡社靜 李濤
?
散發型克雅病7例患者的臨床、腦電圖及影像學分析
郭方亮 胡社靜 李濤
目的 探討散發型克雅病(sCJD)的臨床、腦電圖及影像學特點。方法 回顧性分析7例散發型克雅病患者的臨床表現、腦電圖、影像學特點。結果 本組亞急性起病5例,慢性起病2例,主要的臨床癥狀和體征有進行性癡呆、精神行為異常、視覺障礙、頭暈、共濟失調、肌陣攣、言語笨拙、錐體外系癥狀和錐體束征等;EEG檢查均有異常,其中6例腦電圖檢查示典型的周期性三相波發放,1例患者入院腦電圖檢查未見異常波發放,1月后復查腦電圖發現周期性三相波;7例均行顱腦MRI檢查,T2加權序列(T2WI)、液體衰減反轉恢復序列(T2FLAIR)及彌散加權成像(DWI)在皮質、尾狀核、殼核等發現異常高信號,其中1例在DWI像上發現隨著疾病進展尾狀核、殼核、皮層信號先明顯增高,后稍微下降;6例行腦脊液14-3-3蛋白檢測,其中4例為陽性,2例為陰性。結論 臨床上對快速進展型癡呆的患者,應考慮克雅病的可能,盡早行腦電圖、顱腦MRI以及腦脊液14-3-3蛋白檢測有助于臨床早期診斷;腦電圖、顱腦MRI在疾病早期可無典型改變,則應短期內復查,動態觀察。
散發型克雅病 腦電圖 磁共振成像 14-3-3蛋白
克雅病(Creutzfeldt-Jakob disease,CJD)又被稱為皮質-紋狀體-脊髓變性、亞急性海綿狀腦病或人類傳染性海綿狀腦病,是一種快速進展性、致死性的中樞神經系統變性疾病,也是最常見的一種朊蛋白病(Prion disease),全球范圍內CJD的年發病率為1~2例/百萬。根據病因臨床上常將CJD分為散發型(sporadic CJD, sCJD)、遺傳型、醫源型以及變異型CJD(variant CJD, vCJD),其發病機制是當外來致病朊蛋白(Prion protein, PrP)或遺傳性突變導致正常的PrPc變為PrPsc時PrPsc會促進PrPc轉化為越來越多的PrPsc,致使神經細胞逐漸失去功能,導致神經細胞死亡,最終引起中樞神經系統發生病變。CJD患者常表現為快速進展型癡呆、小腦功能失調、視覺障礙、錐體束征和錐體外系癥狀等多系統損害,其病程短,進展快,致死率100%。支持CJD診斷的輔助檢查結果包括特征性MRI表現、腦電圖上出現周期性同步化三相尖慢波以及腦脊液中14-3-3蛋白陽性[1,2],但都沒達到100%的敏感性和特異性。為探討散發型CJD的臨床、腦電圖(electroencephalography,EEG)及影像學特點,本研究現將近2年本科收治的7例診斷為很可能CJD患者的臨床資料、腦電圖、及影像學特點作一分析。
1 臨床資料
1.1 研究對象 選擇本院神經內科2015年1月~2016年1月收治的7例CJD患者,其中男6例,女1例,年齡49~64歲,平均年齡(57±5)歲;其中來自湖北6例,河南1例;7例中教師2例、職員1例、農民2例、退休2例;均無顱腦手術史、角膜等器官移植史及家族史。
1.2 回顧性分析 詳細采集所有患者的病史,并行體格檢查,總結其臨床表現(表1),全部患者均行腦電圖、顱腦MRI檢查,6例行腰穿檢查取腦脊液標本,1例患者拒絕行腰穿,血清、腦脊液送中國疾病預防控制中心病毒預防控制所行14-3-3蛋白檢測,出院患者進行隨訪。
1.3 臨床表現 5例呈亞急性起病,2例呈慢性起病。首發癥狀頭暈1例,記憶力減退3例、精神癥狀2例,視覺障礙1例。主要臨床癥狀和體征:進行性癡呆7例,精神行為異常4例,共濟失調5例,視物模糊2例,肌陣攣5例,錐體外系癥狀5例,錐體束征1例,言語笨拙2例,癲癇發作1例。
1.4 EEG檢查 7例患者EEG均表現為彌漫性異常腦電活動,對稱或不對稱,其中6例患者首次EEG檢查就顯示各區廣泛慢波背景節律下見周期性尖波、雙相波或三相波間斷發放;另1例患者3月份入院時腦電圖示背景節律減慢、慢波活動增多,未記錄到典型的三相波發放。但在1個多月后復查腦電圖時出現全導周期性雙相波或三相波與彌漫性慢波活動交替出現(圖1)。
1.5 顱腦MRI檢查 7例均見大腦皮質、雙側尾狀核、殼核、基底節等部位異常增高信號,以DWI最為顯著,呈“花邊征”樣改變,T2WI和T2FLAIR也可上述部位信號增高(圖2)。7例T2FLAIR均未見丘腦異常增高信號的“高爾夫球征”。1例除了大腦皮質信號增高,還可見雙側基底節斑片狀高信號(圖3)。其中1例患者分別在2015年5月8日、6月12日、7月21日行顱腦MRI檢查,在DWI像上發現隨著疾病進展尾狀核、殼核、皮層信號先明顯增高,后稍微下降(圖4);值得注意的是,隨疾病進展,該患者在T1FLAIR像上發現腦萎縮進行性加重(圖5)。
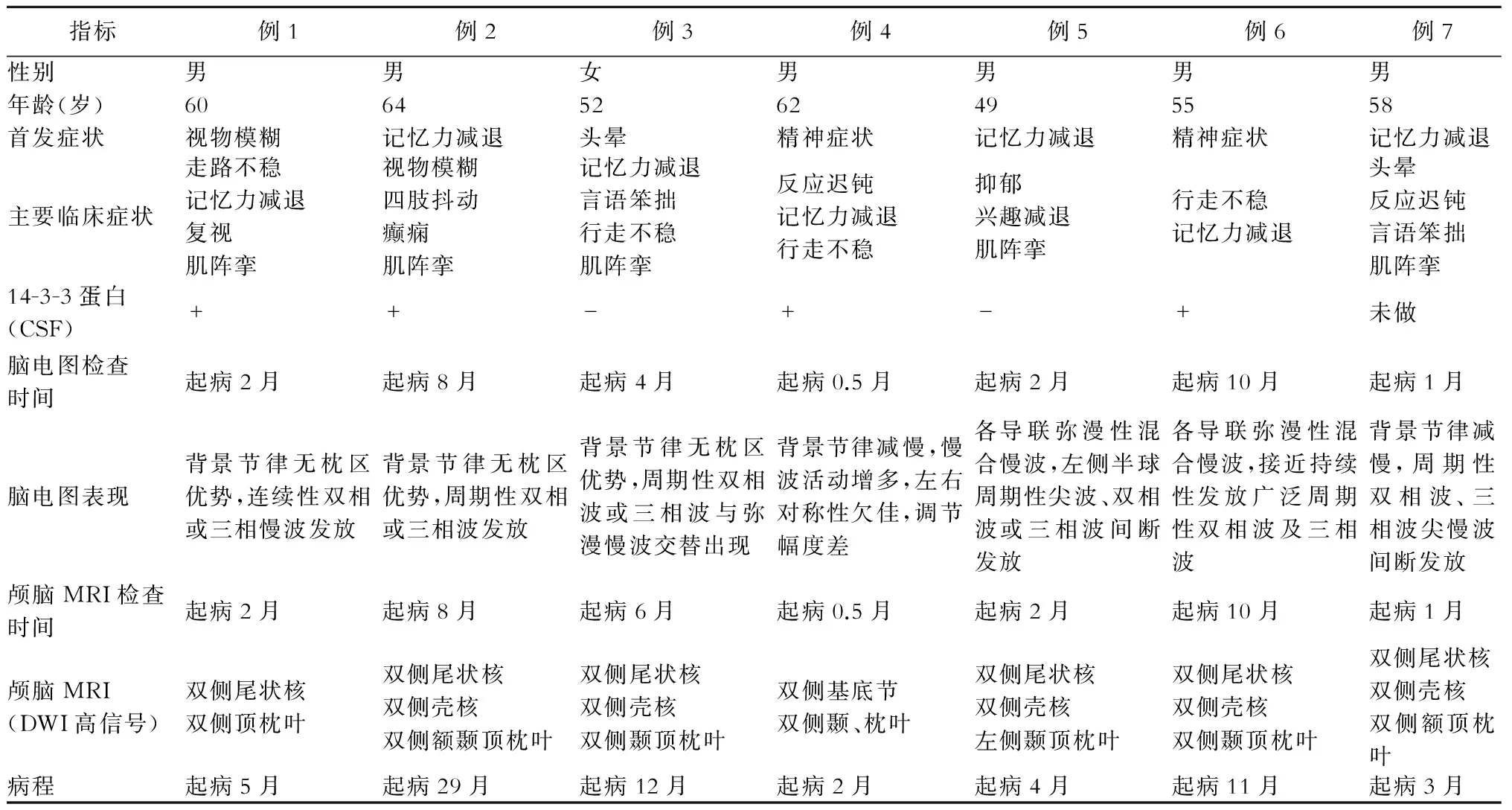
表1 臨床資料
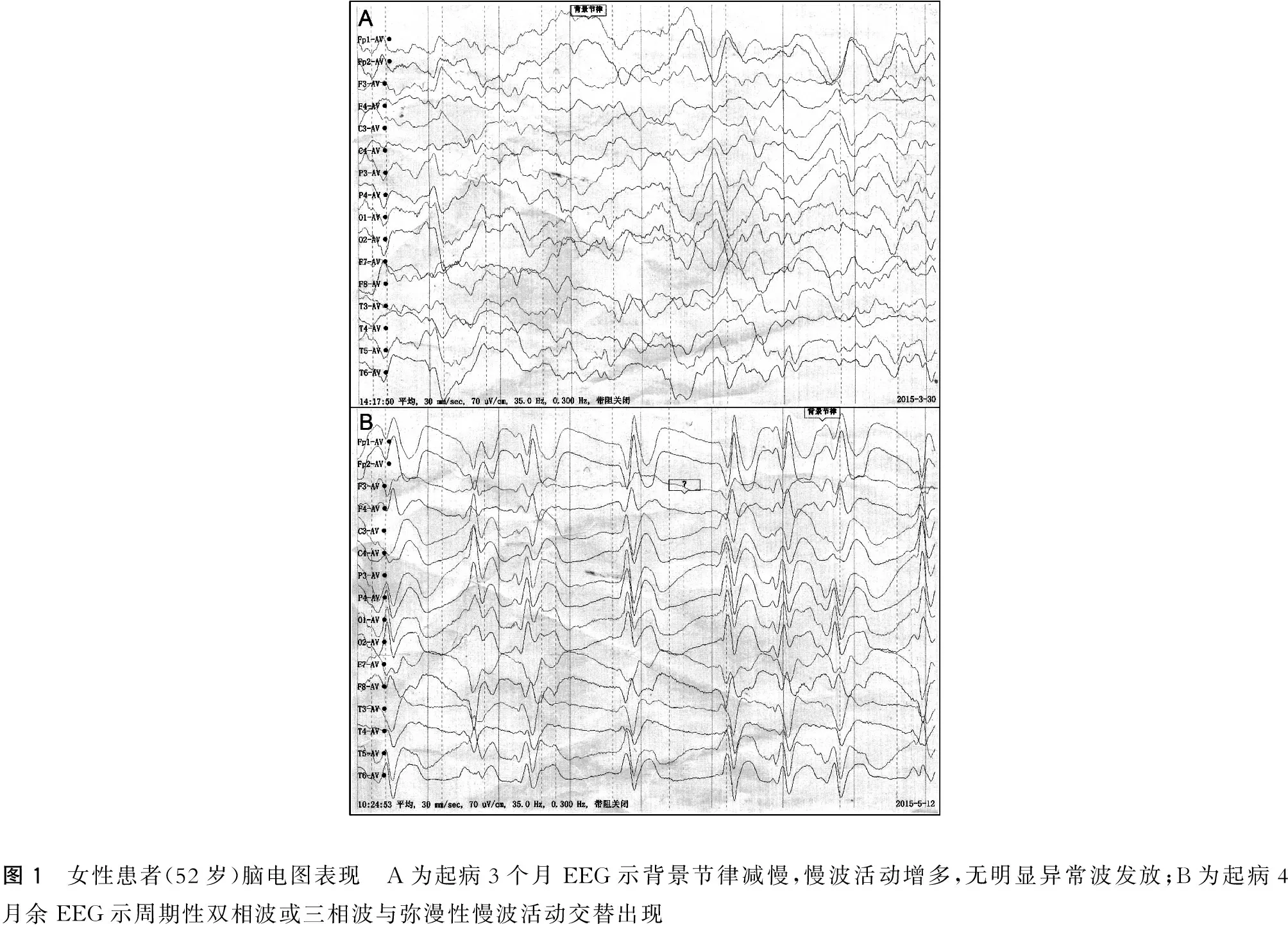
圖1 女性患者(52歲)腦電圖表現 A為起病3個月EEG示背景節律減慢,慢波活動增多,無明顯異常波發放;B為起病4月余EEG示周期性雙相波或三相波與彌漫性慢波活動交替出現
1.6 腦脊液檢查 6例行CSF檢查,其中2例蛋白含量輕度升高,壓力、顏色、有核細胞數正常,培養未見細菌、真菌生長,4例患者14-3-3蛋白(+),2例陰性。
1.7 臨床診斷 7例均診斷為很可能CJD。
2 討 論
CJD最早由Creutzfeld和Jakob先后報道,是一種由內源性或外源性PrP感染所致的可傳染的、致死性的朊蛋白病。WHO將其分為散發型、遺傳型、醫源型、變異型,其中發病率最高的是sCJD,占85%。全球都有sCJD的報道,但沒有地域性和季節性[3-4]。男女均可發病,平均起病年齡在60歲,很少有<40歲或>80歲的人感染sCJD[5-6]。在疾病早期患者可表現為類似于神經衰弱或抑郁癥如情感低落、乏力、失眠、記憶力下降、胃口不佳等癥狀,容易被忽略而未就診或就診于精神科;隨著疾病的進展,則可出現大腦皮質、錐體束、錐體外系等受損的表現如認知功能下降、錐體束征、錐體外系表現、肌陣攣、肌張力升高、震顫、行走不穩等;到疾病晚期則可出現大小便失禁、癡呆、無動性緘默、昏迷甚至去皮質強直狀態。該病進展迅速,其突出特征為快速進展型癡呆和肌陣攣。據統計,從起病到死亡的生存中位期僅為5個月,90%的sCJD患者會在1年內死亡[5,7]。本組的7例患者均考慮為散發型,最突出的癥狀為進行性癡呆(7/7),其次為錐體外系癥狀(5/7),小腦癥狀(5/7);體征方面則包括高級皮層功能障礙、肌張力升高、病理征陽性等。
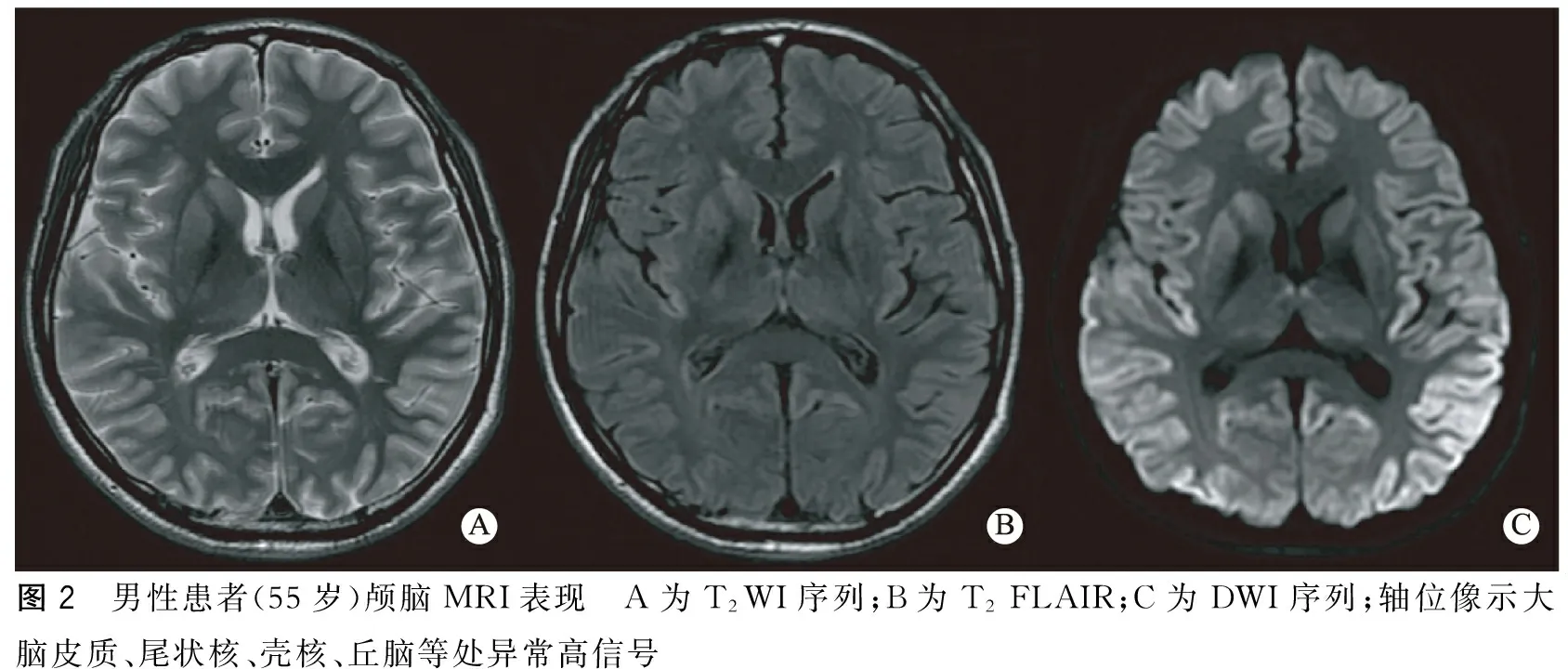
圖2 男性患者(55歲)顱腦MRI表現 A為T2WI序列;B為T2FLAIR;C為DWI序列;軸位像示大腦皮質、尾狀核、殼核、丘腦等處異常高信號
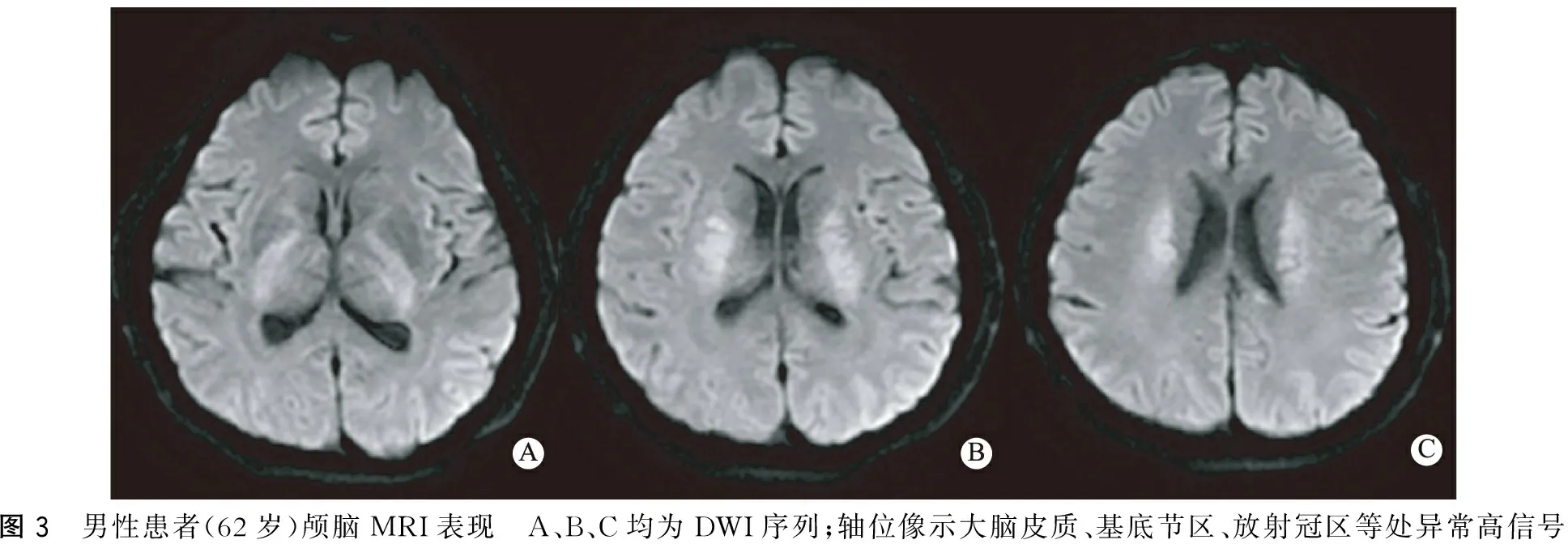
圖3 男性患者(62歲)顱腦MRI表現 A、B、C均為DWI序列;軸位像示大腦皮質、基底節區、放射冠區等處異常高信號
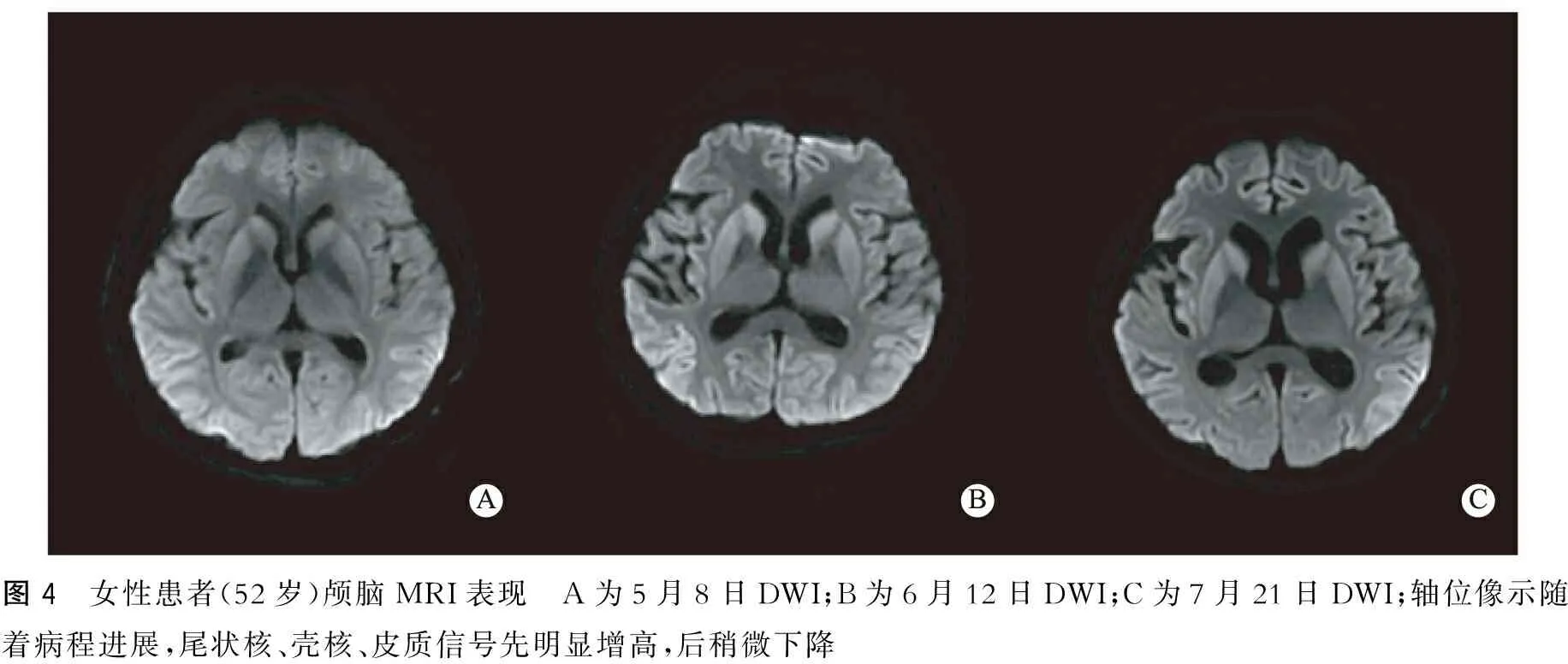
圖4 女性患者(52歲)顱腦MRI表現 A為5月8日DWI;B為6月12日DWI;C為7月21日DWI;軸位像示隨著病程進展,尾狀核、殼核、皮質信號先明顯增高,后稍微下降
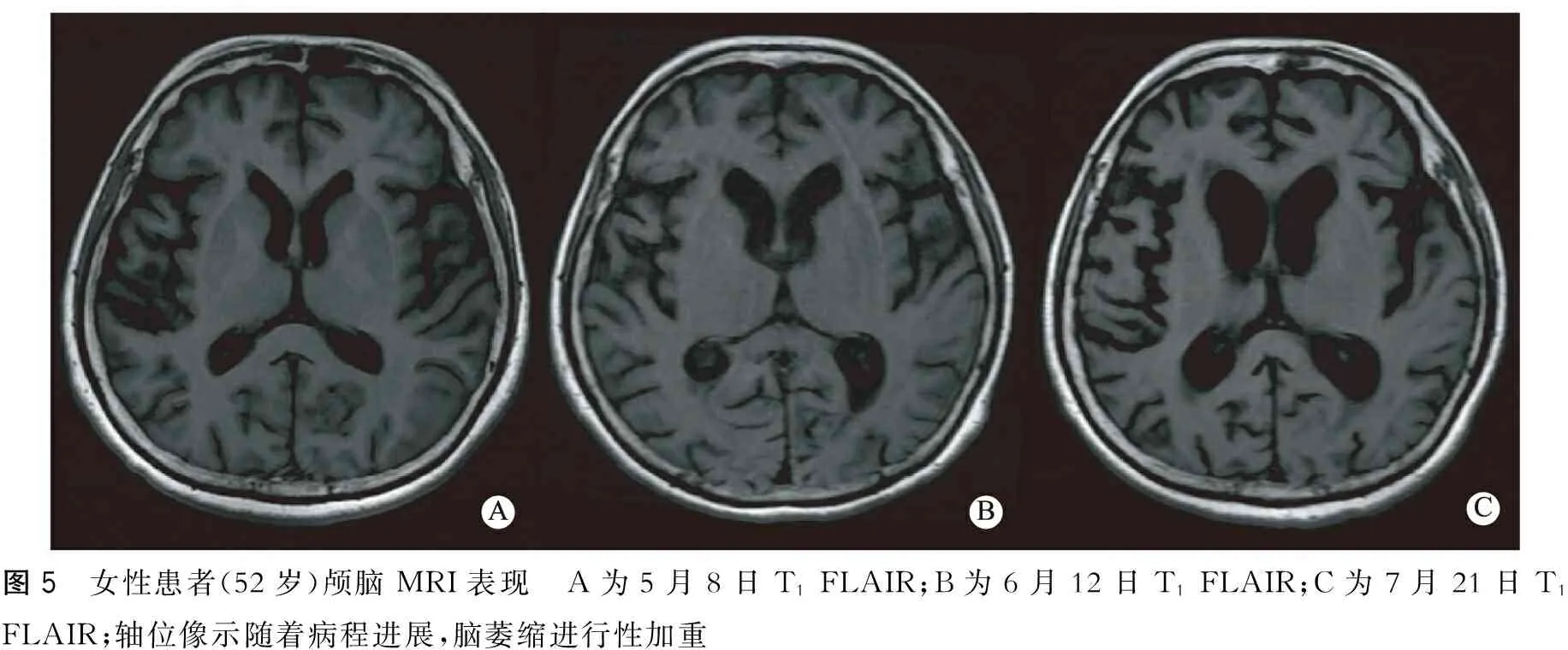
圖5 女性患者(52歲)顱腦MRI表現 A為5月8日T1FLAIR;B為6月12日T1FLAIR;C為7月21日T1FLAIR;軸位像示隨著病程進展,腦萎縮進行性加重
確診CJD需要行組織病理學檢查,電鏡下可發現神經元丟失、神經細胞和樹突中空泡形成,這些病理變化使皮質和腦深部核團呈海綿狀。使用免疫組化染色和western-blot分析可以證實腦組織中含有致病性的朊蛋白[8]。但因活檢為有創檢查,且即便行活檢確診,也沒有有效的治療方法,家屬常難以接受,因此臨床開展很少。目前,許多研究顯示腦電圖、顱腦MRI以及腦脊液14-3-3蛋白的檢測有助于診斷CJD。
腦電圖可反映腦細胞的電活動以及功能狀態。CJD患者早期腦電圖為廣泛存在的非特異性慢波,中后期出現彌漫性慢波(約90%患者可能出現),各導聯間歇性或連續性同步出現中至高波幅,頻率為1~2HZ的三相尖慢波或棘慢波[9],這種特異性周期性同步放電(periodic synchronous discharge)現象是CJD在腦電圖上一個重要的特征性表現,其診斷CJD的特異性為64%,敏感性為91%[1]。有時單次腦電圖檢查并不能顯示這種特征性電活動,可定期復查以提高陽性率。值得注意的是,1例患者入院時腦電圖示背景節律減慢、慢波活動增多,未記錄到典型的三相波發放,但在1月后復查腦電圖發現典型周期性三相波。由此可見,復查腦電圖對于診斷CJD具有重要意義,因為在疾病早期可能不會出現典型的周期性三相波發放。
顱腦MRI是另一項早期診斷CJD的有效方法。目前最新的CJD診斷標準如舊金山加利福尼亞大學、歐洲磁共振-克雅病聯盟推薦的標準都引入了磁共振成像在早期臨床診斷中的作用[10]。顱腦MRI診斷CJD的敏感性為91%,特異性為95%[11],顯示病變的能力以DWI最佳,其次為FLAIR像[2,12],其特征性的表現為DWI、FLAIR、T2WI像上可見大腦皮層、尾狀核、殼核、丘腦等部位出現異常高信號,較少累及的部位包括扣帶回、海馬、島葉和小腦。本組7例患者均行顱腦MRI檢查,所有患者DWI像上均出現大腦皮層異常高信號,呈“花邊征”樣表現,5例患者DWI、T2WI、T2FLAIR像上尾狀核、殼核、丘腦等部位亦可見異常高信號,這些表現都與既往文獻相符。但是,本組7例患者T2FLAIR像均未見雙側丘腦高信號,形成獨特的“高爾夫球征”[13],而這一表現可作為鑒別sCJD和vCJD的重要依據。因此,本組7例患者為散發型的可能性更大。另外,1例患者共行3次顱腦MRI檢查,每次間隔1月余,在DWI像上發現隨著疾病進展尾狀核、殼核、皮層信號先明顯增高,后稍微下降;在T1FLAIR像上發現隨著病程進展腦萎縮進行性加重,這些表現都符合CJD的病理改變。由此可見,復查顱腦MRI尤其是DWI序列也對診斷CJD具有重要意義。因為在疾病早期DWI像上可能不會出現明顯的信號改變,而隨著疾病進展則會出現典型的“花邊征”、基底節高信號等,而當疾病進展到后期時上述部位信號增高的程度可能會較前降低。
14-3-3蛋白是存在于神經細胞中的一種正常信號蛋白,參與許多信號轉導。CJD患者神經細胞大量死亡時14-3-3蛋白被釋放出來,導致CSF中水平明顯升高,但是14-3-3蛋白水平升高也可見于腦梗死、腦炎、阿爾茨海默病等。因此,需排除這些疾病后CSF中14-3-3蛋白水平升高則高度提示CJD。最新的一項研究顯示,腦脊液Tau蛋白水平對克雅病診斷的敏感性和準確性都較14-3-3蛋白水平高[14]。因此,未來的CJD診斷標準可能會引入腦脊液Tau蛋白水平檢測。
目前一項最新的技術被稱為實時誘導轉換(RT-QuIC)能將腦脊液中朊蛋白放大成淀粉樣纖維來檢測,具有80%~90%的敏感性和更高的特異性(99%~100%)[15-16]。Orrú等將該技術用于患者鼻粘膜標本時敏感性和特異性分別為97%和100%[17]。若該技術得到認可,則可能會納入RT-QuIC作為診斷標準的一部分。
診斷CJD需結合臨床特點與輔助檢查,參考2009年歐洲磁共振-克雅病聯盟推薦標準[18]:(1)癡呆、小腦癥狀或視覺障礙、錐體系或錐體外系癥狀、無動性緘默;(2)腦電圖顯示尖慢復合波(Periodic sharp wave complex, PSWC)、腦脊液14-3-3蛋白陽性(起病2年內)、尾狀核和殼核或至少兩個皮質(顳、頂、枕葉)在DWI或FLAIR像上呈異常高信號。滿足(1)中2項和(2)中至少一項診斷很可能CJD;滿足(1)中2項并且病程小于2年診斷為可能CJD;腦組織活檢發現海綿狀變性和PrPsc可確診CJD。本組7例患者均未做活檢,根據診斷標準7例均診斷為很可能CJD。
在臨床工作中對于中老年患者出現快速進展型癡呆伴有肌陣攣、共濟失調、精神行為異常、錐體束征、錐體外系表現等多系統受累時要考慮CJD的可能,應盡早行腦電圖、顱腦MRI(尤其是DWI、T2FLAIR)以及腰穿來幫助診斷。若早期腦電圖、顱腦MRI未見典型改變,則應定期復查,以發現提示CJD的特征性改變,并為動態觀察疾病進展提供客觀依據。
[1] Steinhoff BJ,Zerr I,Glatting M,et al.Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease[J].Ann Neurol,2004,56(5):702-708.
[2] Tschampa HJ,Kallenberg K,Urbach H,et al.MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease: a study on inter-observer agreement[J].Brain,2005,128(Pt 9):2026-2033.
[3] Ladogana A,Puopolo M,Croes EA,et al.Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada[J].Neurology,2005,64(9):1586-1591.
[4] Linsell L,Cousens SN,Smith PG,et al.A case-control study of sporadic Creutzfeldt-Jakob disease in the United Kingdom: analysis of clustering[J].Neurology,2004,63(11):2077-2083.
[5] Brown P,Gibbs CJ,Rodgers-Johnson P,et al.Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease[J].Ann Neurol,1994,35(5):513-529.
[6] Will RG,Matthews WB.A retrospective study of Creutzfeldt-Jakob disease in England and Wales 1970-79. I: Clinical features[J].J Neurol Neurosurg Psychiatry,1984,47(2):134-140.
[7] Johnson RT,Gibbs CJ.Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies[J].N Engl J Med,1998,339(27):1994-2004.
[8] Johnson RT.Prion diseases[J].Lancet Neurol,2005,4(10):635-642.
[9] 黃勛,范學工.克雅病研究進展[J].臨床內科雜志,2010,27(5):293-295.
[10]Newey CR,Sarwal A,Wisco D,et al.Variability in diagnosing Creutzfeldt-Jakob disease using standard and proposed diagnostic criteria[J].J Neuroimaging,2013,23(1):58-63.
[11]Young GS,Geschwind MD,Fischbein NJ,et al.Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis[J].AJNR Am J Neuroradiol,2005,26(6):1551-1562.
[12]Fragoso DC,Gon alves Filho AL,Pacheco FT,et al.Imaging of Creutzfeldt-Jakob disease: imaging patterns and their differential diagnosis[J].Radiographics,2017,37(1):234-257.
[13]Collie DA,Summers DM,Sellar RJ,et al.Diagnosing variant Creutzfeldt-Jakob disease with the pulvinar sign: Mr imaging findings in 86 neuropathologically confirmed cases[J].AJNR Am J Neuroradiol,2003,24(8):1560-1569.
[14]Hamlin C,Puoti G,Berri S,et al.A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease[J].Neurology,2012,79(6):547-552.
[15]Kim MO,Geschwind MD.Clinical update of Jakob-Creutzfeldt disease[J].Curr Opin Neurol,2015,28(3):302-310.
[16]Manix M,Kalakoti P,Henry M,et al.Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy[J].Neurosurg Focus,2015,39(5):E2.
[17]Orrú CD,Bongianni M,Tonoli G,et al.A test for Creutzfeldt-Jakob disease using nasal brushings[J].N Engl J Med,2014,371(6):519-529.
[18]Zerr I,Kallenberg K,Summers DM,et al.Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease[J].Brain,2009,132(Pt 10):2659-2668.
(2016-09-22收稿)
The study on clinical feature, electroencephalography and MRI in seven cases of sporadic Creutzfeldt-Jakob disease
Guo Fangliang, Hu Shejing, Li Tao.
Department of Neurology, Renmin Hospital of Wuhan University, Wuhan 430060
Objective To investigate the clinical feature, electroencephalography (EEG) and MRI of sporadic Creutzfeldt-Jakob disease (sCJD).Methods The clinical feature, EEG and MRI of 7cases of sCJD were analyzed retrospectively.Results 5 cases were subacute onset, 2 cases were chronic onset. The major clinical symptoms and signs were progressive dementia, mental and behavior disorder, visual abnormalities, dizziness, ataxia, myoclonus, speech clumsy, extrapyramidal symptoms and pyramidal sign etc. The EEG results were all abnormal, and 6 cases showed typical periodic triphasic wave. One patient showed no abnormal waves when admitted to hospital, but showed periodic triphasic wave on reexamination of EEG one month later. Head magnetic resonance imaging (MRI) was performed in all cases and showed hyperintense signals in cerebral cortex, caudate nucleus and putamen on T2-weighted image (T2WI), T2fluid-attenuated inversion recovery image (T2FLAIR) and diffusion-weighted image (DWI). One patient showed the hyperintense signals in cerebral cortex, caudate nucleus and putamen enhanced first and then attenuated with the progression of the disease. Detection of 14-3-3 protein in cerebrospinal fluid was performed in 6 cases and 4 cases showed positive results.Conclusion Patients with rapidly progressive dementia should be taken into consideration of CJD. Examination with EEG, head MRI and detection of 14-3-3 protein are helpful for establishing the early diagnosis in suspected cases. Sometimes no typical findings of CJD were found on EEG and head MRI in early stage, reexamination of EEG and MRI should be performed in a short time.
Sporadic Creutzfeldt-Jakob disease Electroencephalography MRI 14-3-3 protein
430060 武漢大學人民醫院神經內科[郭方亮 胡社靜 李濤(通信作者)]
R742
A
1007-0478(2017)03-0217-06
10.3969/j.issn.1007-0478.2017.03.012
猜你喜歡
現代儀器與醫療(2021年4期)2021-11-05 08:25:32
皮膚病與性病(2021年3期)2021-07-30 08:07:32
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
科技傳播(2019年22期)2020-01-14 03:06:40
中國生殖健康(2019年3期)2019-02-01 06:12:26
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
疑難病雜志(2014年12期)2014-04-16 05:19:34
河南科技(2014年22期)2014-02-27 14:18:22
