晚發型脊柱骨骺發育不良伴進行性骨關節病1家系報道及基因研究
2017-08-07 06:56:52劉書中李政垚李子全王牧川王以朋
中國實驗診斷學 2017年7期
關鍵詞:基因突變
劉書中,宋 桉,李政垚,李子全,王牧川,王以朋*
(中國醫學科學院 北京協和醫學院 北京協和醫院 1.骨科;2.內分泌科,北京100730)
*通訊作者
晚發型脊柱骨骺發育不良伴進行性骨關節病1家系報道及基因研究
劉書中1,宋 桉2,李政垚1,李子全1,王牧川1,王以朋1*
(中國醫學科學院 北京協和醫學院 北京協和醫院 1.骨科;2.內分泌科,北京100730)
晚發型脊柱骨骺發育不良(spondyloepiphyseal dysplasia,SED)系一組同時累及脊柱和長管狀骨骨骺的遺傳性發育障礙性疾病,臨床上有軀干與肢體不成比例的矮小身材特征。晚發型SED發病率低,約為0.1-0.4/10萬,是一種較為罕見的骨發育不良疾病[1]。
1 臨床資料
患者,女,31歲,因間斷性全身多關節疼痛25年、加重5年就診。該患者6歲前發育正常,6歲后無明顯誘因出現雙側髖部疼痛,程度逐漸加重,并逐漸出現雙膝、雙肘、雙側指間關節、雙腕、雙肩疼痛及活動受限,發育較同齡人遲緩,病
程中無晨僵,無腰背痛、足跟痛,無腹痛、腹瀉、尿頻、尿急、眼部不適等表現。查體:身高145 cm,智力正常,短頸,肩峰高聳,桶狀胸。脊柱前屈、后伸、左右側屈均明顯受限,雙髖關節內旋受限,雙側肩、肘、腕關節活動明顯受限,雙肘關節屈曲畸形,爪形手,雙側骶髂關節壓痛(+),雙側4字試驗(+)。實驗室檢查:血尿便常規、紅細胞沉降率、C反應蛋白、肝腎功、電解質、凝血功能均無顯著異常,免疫球蛋白、補體、RF、ASO、HLA-B27、ACL、ANA、抗CCP抗體均(-)。脊柱正側位X線:椎體橫徑增大,兩側緣低平,中央部膨突,見圖1。四肢長骨X線檢查:長管狀骨明顯短縮,干骺端擴張,結構紊亂,不均勻致密,邊緣呈刺狀突出。骨骺形態不規則,邊緣不規整,部分呈碎裂狀,脛骨兩側向內傾斜,呈“O”形改變。雙手正位:雙側指間關節間隙狹窄,骨性膨大,屈曲畸形。骨盆正位:雙髖關節間隙狹窄,股骨頭塌陷變形,見圖2。雙膝X線:雙膝關節間隙狹窄,骨質增生。家系調查:家族中無近親結婚,母親孕期無患病或服藥史。進一步對該患者家系進行了詳細調查,繪制了家系圖譜(見圖3),該家系3代中2例患者均為女性,先證者為該患者姐姐,發病年齡均在6-8歲之間。
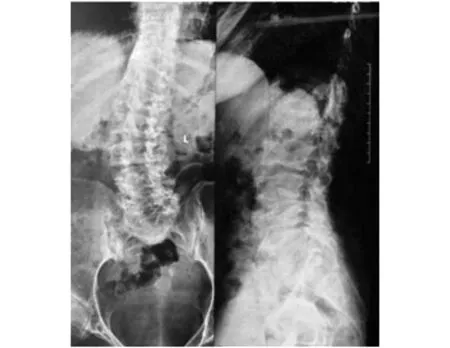
圖1 先證者脊柱正側位X線片
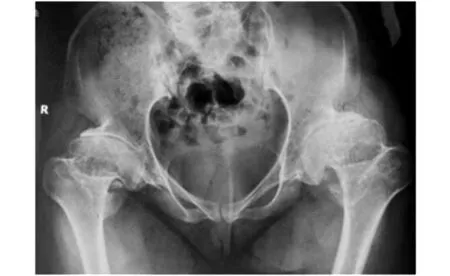
圖2 先證者骨盆正位X線片
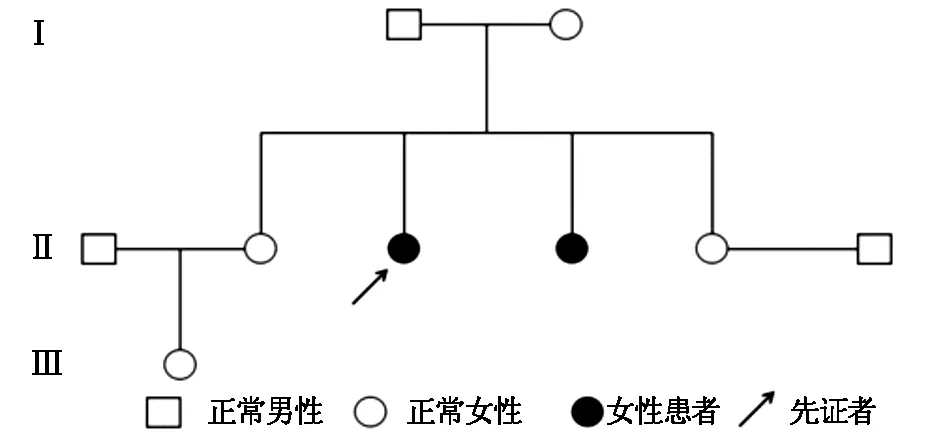
圖3 患者家系遺傳圖譜
2 對象和方法
經患者及家屬知情同意后,采集家系成員外周血6份(10 ml/人),同時抽取30名無關志愿者靜脈血液進行對照試驗,其中男10名,女20名。之后采用TIANamp Blood DNA Kit提取基因組DNA,記錄DNA濃度后,EP管密封-4℃保存備用。設計引物擴增WISP3等363個基因。擴增步驟為:預變性溫度為94℃ 3分鐘,94℃ 55秒,64℃ 55秒,72℃ 60秒,35個循環,最后延伸為72℃ 10分鐘,PCR產物用1.5%瓊脂糖凝膠電泳分離,溴乙錠染色后觀察條帶。使用相同的引物,測序儀器為ABI PRISM 3730,測序試劑為BigDye terminator v3.1,由天津華大基因生物公司測序部對PCR擴增產物進行純化及測序。將突變片段的測序結果與GeneBank中363個基因外顯子DNA序列進行比較。
3 結果
基因檢測檢出該家系中兩位患者均攜帶WISP3基因的2個雜合突變c.396T>G(p.Cys132Trp)和c.721T>G(p.Cys241Gly),c.396T>G(p.Cys132Trp)和c.721T>G(p.Cys241Gly)均未經文獻報道,可分別導致WISP3基因編碼的第132位半胱氨酸突變為色氨酸以及第241位半胱氨酸突變為甘氨酸。在家系中正常個體及30名對照無關志愿者中未發現上述兩個基因突變存在。
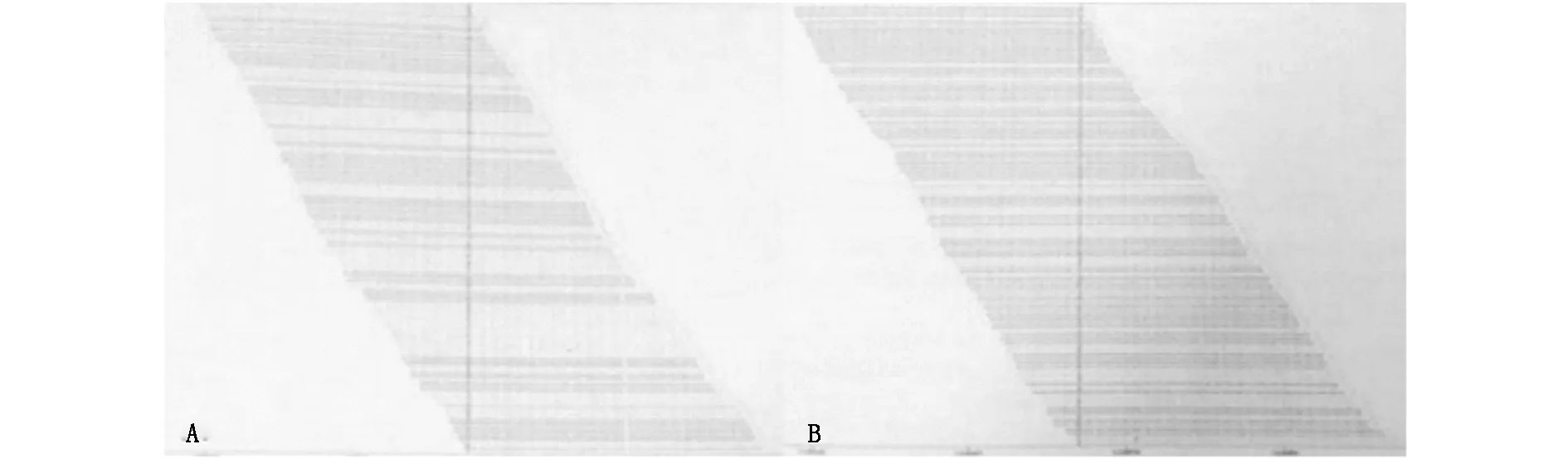
圖4 (A)WISP3基因突變c.396T>G(p.Cys132Trp)支持圖。(B)WISP3基因突變c.721T>G(p.Cys241Gly)支持圖
4 討論
SED根據發病時間不同可分為早發型(先天型)和晚發型(遲發型)兩種[2]。早發型SED系常染色體顯性遺傳病,患兒出生后即見異常。晚發型SED患者通常在5歲以后出現癥狀,其遺傳方式包括X染色體隱性遺傳(僅男性發病)或常染色體顯性及隱性遺傳(男女均可發病),本型在臨床上主要以短軀干侏儒、腰部與四肢大關節疼痛及活動受限為特征。本型主要X線特征包括扁平椎、椎間隙狹窄或增寬、脊柱前側凸、髖內翻畸形、股骨遠端及肱骨近端干骺端變寬而不規則并可伴有骨贅形成等[3]。隨病變進展,全身多關節疼痛及活動受限逐漸加重,本家系中2例患者臨床特點均與之相符。
臨床上晚發型SED需與以下疾病進行鑒別:①黏多糖病IV型:是常染色體隱性遺傳疾病,發病常與近親結婚有關,此外尚有肝脾腫大、角膜混濁、耳聾、尿黏多糖呈強陽性等特異表現,X線中“子彈頭樣椎體”和“飄帶樣肋骨”是其特征性影像學改變,尿篩查試驗及血酶活性測定可予鑒別[4]。②Scheuermann病(椎體骺板缺血性壞死):常在10歲左右發病,可侵犯全部胸腰椎,椎體前緣楔形變窄,典型者呈階梯狀,但無骨盆及椎間隙狹窄改變[5]。此外,還需要進行鑒別的疾病包括成骨不全綜合征、脊柱干骺端發育不良、多發性骨骺發育不良及幼年型類風濕關節炎等。
診斷方面,晚發型SED診斷的確立需結合患者發病特征、臨床表現、特征影像學改變以及基因測序,在疾病問診中需特別注意對家族史的詢問。該家系中兩位患者均攜帶WISP3基因的2個雜合突變c.396T>G(p.Cys132Trp)和c.721T>G(p.Cys241Gly),二者均未經文獻報道,可分別導致WISP3基因編碼的第132位半胱氨酸突變為色氨酸以及第241位半胱氨酸突變為甘氨酸,而在家系中正常個體及30名對照無關志愿者中未發現上述兩個基因突變存在,WISP3相關的晚發型脊柱骨骺發育不良伴進行性骨關節病為常染色體隱性遺傳,純合或復合雜合基因突變可導致疾病發生,因此,c.396T>G(p.Cys132Trp)和c.721T>G(p.Cys241Gly)突變很可能形成復合雜合形式,進而與致病相關[6-8]。
治療方面,目前針對晚發型SED尚缺乏有效的治療手段。對于晚發型SED患者,在疾病早期可予適當抗骨質疏松、營養軟骨、消炎鎮痛治療,但長期療效有待進一步論證。對于晚期SED患者出現全身嚴重畸形及功能障礙時,為提高患者生活質量、改善畸形,可酌情行假體置換或人工關節置換術[9]。近年來,晚發型SED相關致病基因位點不斷被發現,使得在分子水平對晚發型SED進行診斷及治療成為可能,但尚需進一步深入研究晚發型SED的遺傳特點,以期將新的診療技術早日應用于臨床造福該病患者。
[1]GedeonAK,ColleyA,Jamieson R,et al.Identification of the gene(SEDL) causing X-linked spondyloepiphysealdysplasia tarda[J].NatGenet,1999,22(4):400.
[2]Kocyigit H,Arkun R,Ozkinay F,et al.Spondyloepiphyseal dysplasia tarda with progressive arthropathy[J].Clin Rheumatol,?2000,19(3):238.
[3]張 華,杜聯軍,張 歡,等.脊柱骨骺發育不良的影像學表現[J].診斷學理論與實踐,2004,3(3):182.
[4]楊 波,金 今,翁習生,等.6例脊柱骨骺發育不良患者的診斷及治療[J].中國骨與關節外科,2008,1(4):285.
[5]孫 靜,夏維波,李 梅,等.遲發性脊柱骨骺發育不良的臨床診斷及SEDL基因突變分析[J].中華骨質疏松和骨礦鹽疾病雜志,2012,5(1):7.
[6]Liu L,Li N,Zhao Z,et al.Novel WISP3 mutations causing spondyloepiphyseal dysplasiatardawithprogressive arthropathy in two unrelated Chinese families[J].Joint Bone Spine,2015,82(2):125.
[7]Jurgens J,Sobreira N,Modaff P,et al.Novel COL2A1 variant (c.619G>A,p.Gly207Arg) manifestingas a phenotype similar to progressivepseudorheumatoid dysplasiaandspondyloepiphysealdysplasia,Stanescu type[J].Hum Mutat,2015,36(10):1004.
[8]Montané LS,Marín OR,Rivera-Pedroza CI,et al.Early severe scoliosis in a patient with atypicalprogressive pseudorheumatoid dysplasia(PPD):Identification of two WISP3 mutations,one previously unreported[J].Am J Med Genet A,2016,170(6):1595.
[9]Kim RH,Scuderi GR,Dennis DA,et al.Technical challenges of total knee arthroplasty inskeletaldysplasia[J].Clin Orthop Relat Res,2011,469(1):69.
1007-4287(2017)07-1225-03
2016-07-24)
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
