4例以黃疸為主要表現(xiàn)的紅細(xì)胞生成性原卟啉病臨床、病理及遺傳學(xué)分析
2017-08-07 10:05:38李曉青賴雅敏張博為武麗娜朱麗明錢家鳴
臨床肝膽病雜志 2017年7期
關(guān)鍵詞:肝功能
李曉青, 師 杰, 賴雅敏, 張博為, 武麗娜, 朱麗明, 張 濤, 錢家鳴
(中國醫(yī)學(xué)科學(xué)院, 北京協(xié)和醫(yī)學(xué)院, 北京協(xié)和醫(yī)院 a.消化內(nèi)科; b.病理科;c.內(nèi)科; d.皮膚科, 北京 100730)
4例以黃疸為主要表現(xiàn)的紅細(xì)胞生成性原卟啉病臨床、病理及遺傳學(xué)分析
李曉青a, 師 杰b, 賴雅敏a, 張博為c, 武麗娜a, 朱麗明a, 張 濤d, 錢家鳴a
(中國醫(yī)學(xué)科學(xué)院, 北京協(xié)和醫(yī)學(xué)院, 北京協(xié)和醫(yī)院 a.消化內(nèi)科; b.病理科;c.內(nèi)科; d.皮膚科, 北京 100730)
目的 探討紅細(xì)胞生成性原卟啉病的臨床、病理及遺傳學(xué)特點(diǎn),以期提高對該病肝臟受累的認(rèn)識。方法 回顧性分析2011年7月-2014年9月在北京協(xié)和醫(yī)院住院的4例以黃疸為主要表現(xiàn)的紅細(xì)胞生成性原卟啉病患者的臨床特征、肝組織病理以及突變基因特點(diǎn)。結(jié)果 4例患者均以急性/亞急性起病,肝內(nèi)膽汁淤積性黃疸為主要臨床特征,肝功能:GGT 425~1152 U/L,ALP 196~356 U/L,TBil 287~485 μmol/L,DBil 216~394 μmol/L。追溯4例患者年幼即出現(xiàn)典型日照后皮膚疼痛、紅斑、水皰。進(jìn)一步檢查紅細(xì)胞游離原卟啉49.8~113.1 μg/gHb,肝臟組織病理在偏光顯微鏡下均可見“Maltese”十字,并檢測到FECH基因不同位點(diǎn)的突變。結(jié)論 對于肝內(nèi)膽汁淤積性肝病合并典型日照后痛性紅斑,應(yīng)警惕紅細(xì)胞生成性原卟啉病,皮膚/肝臟病理、紅細(xì)胞內(nèi)原卟啉、FECH基因檢測有助于該病的診斷。
原卟啉病, 紅細(xì)胞生成性; 黃疸; 皮炎, 光變態(tài)反應(yīng)
卟啉病是由于血紅素生物合成途徑中酶的缺乏引起卟啉或其前體濃度異常升高,并在組織中蓄積的一組疾病,包括遺傳性和獲得性卟啉病。其代謝途徑中8種酶的缺陷對應(yīng)著8種卟啉病,急性間歇性卟啉病、遲發(fā)性皮膚卟啉病和紅細(xì)胞生成性原卟啉病(erythropoietic protoporphyria, EPP)是最常見的3種類型。EPP是一種常染色體顯性遺傳病,突變基因是亞鐵螯合酶(ferrochelatase,F(xiàn)ECH)基因,導(dǎo)致線粒體鐵螯合酶的缺陷,引起原卟啉在紅細(xì)胞、血漿、肝臟和皮膚的蓄積,出現(xiàn)相應(yīng)的臨床癥狀。EPP屬罕見疾病,其發(fā)病率為1/75 000~1/200 000[1],主要表現(xiàn)是幼兒時期即出現(xiàn)的痛性皮膚光過敏伴紅斑。肝膽系統(tǒng)受累不多見,發(fā)生在1%~4%EPP患者中,表現(xiàn)為膽汁淤積和慢性肝病,嚴(yán)重者可導(dǎo)致肝功能衰竭。本文總結(jié)了4例以肝內(nèi)膽汁淤積表現(xiàn)為主的EPP。
1 資料及方法
檢索北京協(xié)和醫(yī)院住院患者中診斷為“紅細(xì)胞生成性原卟啉病”的患者,住院時間為2011年7月-2014年9月,共收集4例。EPP的診斷依靠臨床表現(xiàn)、肝臟病理以及血紅細(xì)胞游離原卟啉顯著升高。回顧性分析這4例EPP患者的臨床表現(xiàn)、肝臟組織病理及基因遺傳學(xué)特點(diǎn)。
2 結(jié)果
2.1 人口學(xué)特征 4例EPP患者中男2例,女2例,就診年齡16~45歲。
2.2 臨床表現(xiàn)及特點(diǎn)
2.2.1 黃疸 4例患者均以黃疸為主要表現(xiàn)就診,黃疸病程20天~5月,表現(xiàn)為尿色加深、皮膚及鞏膜黃染,伴有乏力、惡心、納差。3例就診于消化內(nèi)科,1例就診于急診科。
2.2.2 腹痛 4例患者中3例出現(xiàn)明顯腹痛,表現(xiàn)為持續(xù)全腹痛、腹脹、排氣排便減少,立位腹平片提示腸梗阻,但胃鏡、結(jié)腸鏡、消化道造影、腹盆CT未發(fā)現(xiàn)機(jī)械性梗阻證據(jù)。
2.2.3 皮疹 4例患者均可追溯到自幼(3~4歲開始)即出現(xiàn)光照5~30 min后皮膚出現(xiàn)明顯燒灼感和疼痛,隨即皮膚出現(xiàn)紅斑和水腫,部分有水皰、結(jié)痂。但患者在皮膚科就診中多次被診斷為“日光性皮炎”,未予重視。
2.2.4 家族史 4例患者中1例有卟啉病家族史(患1),其他3例患者無明確家族史。
2.3 實驗室檢查及影像學(xué)結(jié)果
2.3.1 血常規(guī) WBC(4.3~12.8)×109/L,Hb(88~105) g/L,2例患者PLT(28~34)×109/L,另2例患者PLT(106~173)×109/L。
2.3.2 肝功能及卟啉相關(guān)檢查結(jié)果 4例EPP患者肝功能(包括ALT、AST、GGT、ALP、TBil和DBil)及卟啉相關(guān)檢查結(jié)果(包括紅細(xì)胞游離原卟啉、尿卟啉和尿卟膽原)見表1,其中尿卟啉及尿卟膽原均提示陰性。
2.3.3 病毒性肝炎指標(biāo) HAV-IgM、HBsAg、抗-HCV、HEV-IgM、巨細(xì)胞病毒DNA(CMV-DNA)、EB病毒DNA(EBV-DNA)均陰性。
2.3.4 免疫指標(biāo) 抗核抗體(ANA)、抗線粒體抗體(AMA)、AMA-M2、抗平滑肌抗體(SMA)均陰性。
2.3.5 腹部超聲檢查/CT 肝脾輕度腫大,無肝內(nèi)外膽管擴(kuò)張表現(xiàn)。1例患者(患4)CT提示肝大、巨脾、腹盆腔大量積液。
2.4 肝臟病理 因患者均以黃疸為主要表現(xiàn)就診,在除外肝外梗阻后,4例患者均創(chuàng)造條件做了經(jīng)皮肝臟穿刺。肝組織病理HE染色(圖1)顯示肝實質(zhì)內(nèi)彌漫毛細(xì)膽管擴(kuò)張、充滿棕色粗大管型,部分管型脫落于肝竇內(nèi),肝細(xì)胞漿內(nèi)亦可見較多棕色顆粒沉積;部分肝細(xì)胞明顯腫脹呈氣球樣變,伴小泡性脂變,部分肝細(xì)胞呈大泡性脂變。偏光鏡下這些管型或顆粒呈鮮紅色雙折光,其中可見Maltese十字(圖2)。
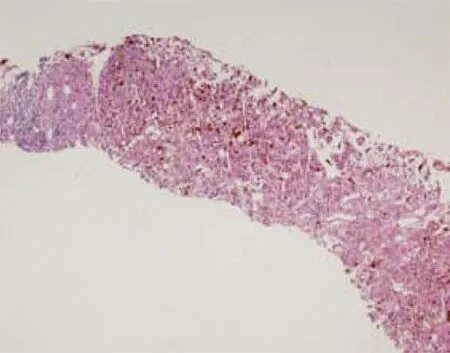
圖1 肝臟穿刺病理(HE染色,×4)
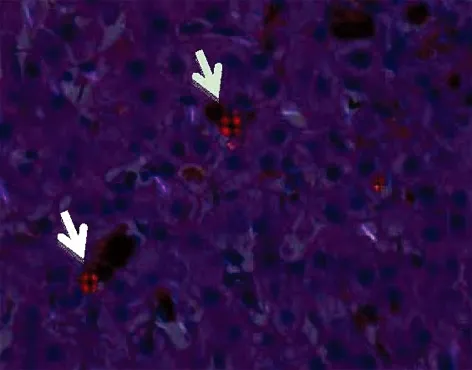
圖2 偏光顯微鏡結(jié)果(×200) 白色箭頭所指為“Maltese”十字
2.5 基因檢測 4例患者均進(jìn)行了FECH基因檢測,檢測到FECH基因不同位點(diǎn)的突變;對部分家系進(jìn)行篩查,其中3例檢測到與患者相同的突變位點(diǎn)(表2)。
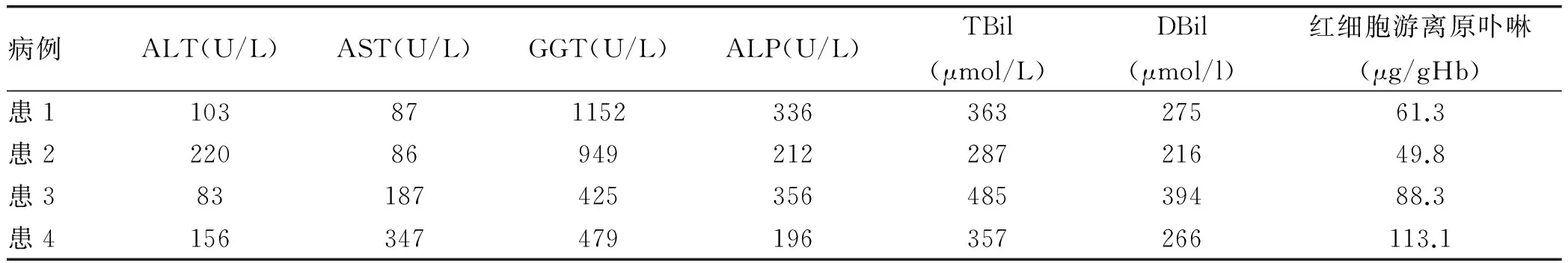
表1 4例EPP患者肝功能及卟啉相關(guān)檢查結(jié)果
注:紅細(xì)胞游離原卟啉正常值0~4.7 μg/gHb
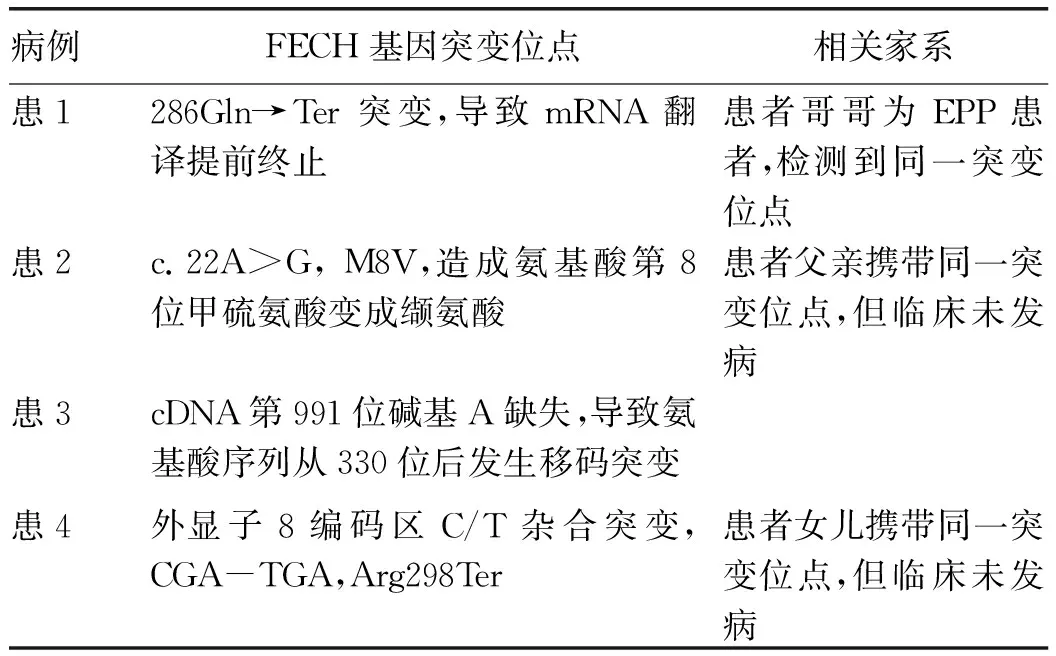
表2 EPP患者及部分家系基因檢測結(jié)果
2.6 治療及預(yù)后 4例患者以黃疸為主要表現(xiàn)入院,明確診斷EPP后,因針對肝臟病變無特異性治療方案,予以熊去氧膽酸等保肝治療,3例患者短時間內(nèi)應(yīng)用糖皮質(zhì)激素減輕淤膽,4例患者膽紅素及GGT、ALP水平略有下降,出院隨訪。針對皮膚病變建議患者避光,并服用β胡蘿卜素。1年后隨訪,4例患者仍存在中度以上黃疸,TBil 100~200 μmol/L,服用考來烯胺、熊去氧膽酸等保肝藥物及中藥治療中。
3 討論
EPP是一種遺傳性疾病,最早在1961年Magness等報告了EPP病例。大部分EPP患者是染色體18q21.3-22區(qū)域基因異常導(dǎo)致FECH部分缺失所致。FECH是卟啉代謝過程中的第8個酶,催化鐵與光卟啉IX結(jié)合形成血紅素。EPP患者FECH的活性僅為正常人的10%~25%,F(xiàn)ECH的缺失使原卟啉大量沉積在紅細(xì)胞內(nèi),隨著紅細(xì)胞的老化釋放到血漿中。原卟啉可由肝排入膽汁和糞便中,因此EPP患者紅細(xì)胞、血漿和大便中原卟啉增加,而尿卟啉陰性,這是EPP的特點(diǎn)。
EPP典型的皮膚表現(xiàn)是患者常在幼年(3~5歲)出現(xiàn)日曬后暴露部位急性燒灼樣疼痛,伴有紅斑腫脹,有時出現(xiàn)紫癜、丘疹、水皰和血皰,繼而潰爛結(jié)痂,反復(fù)日曬部位如手背、關(guān)節(jié)骨突皮紋加深,呈蠟樣增厚;部分患者口周有放射性萎縮性紋理,稱假性皸裂。皮膚病理表現(xiàn)為表皮萎縮,棘層細(xì)胞內(nèi)可見較多空泡化細(xì)胞,基底層點(diǎn)狀液化變性,真皮淺中層血管和膠原纖維之間可見淋巴細(xì)胞、組織細(xì)胞浸潤,血管周圍纖維素樣物質(zhì)和毛玻璃樣PAS陽性物質(zhì)沉積于血管周圍,血管壁和基底層增厚[2]。國內(nèi)文獻(xiàn)報道的EPP病例以皮膚表現(xiàn)為主[2-6],就診皮膚科的患者需要與牛痘樣水皰病、著色性干皮病、日光性蕁麻疹以及其他類型的卟啉病相鑒別。典型的皮膚表現(xiàn)和病理,結(jié)合紅細(xì)胞、血漿和糞便中原卟啉升高可以協(xié)助EPP診斷。控制光敏感是EPP皮膚病變的基礎(chǔ)治療,患者需要通過衣物和遮光劑如二氧化鈦和二氧化鋅來避免陽光照射。β胡蘿卜素用于增強(qiáng)患者對日光的耐受,對大部分患者起到保護(hù)作用,推薦劑量每日口服60~180 mg,維持血清濃度在4~6 mg/L。補(bǔ)骨脂素長波紫外線可增加真皮厚度和表皮黑素,對EPP皮膚病變可能有幫助。
本文報告的4例患者自幼即有光敏感癥狀,也表現(xiàn)為典型的光照后燒灼痛、紅斑、水皰及結(jié)痂,且有1例患者有明確卟啉病家族史,但在出現(xiàn)黃疸前這部分患者并沒有診斷EPP,提示皮膚科醫(yī)師應(yīng)提高對EPP典型皮疹的認(rèn)識,早期診斷、早期干預(yù)、早期監(jiān)測。
肝膽系統(tǒng)受累在EPP患者中并不常見,當(dāng)卟啉生成過多超過肝臟代謝時,卟啉在肝臟淤積形成膽結(jié)石,并出現(xiàn)肝功能異常,1%~4%嚴(yán)重者引起肝硬化、肝功能衰竭。當(dāng)患者出現(xiàn)肝臟或膽道癥狀時,皮膚癥狀常會被忽視或被認(rèn)為與此病無關(guān),而造成疾病的漏診、誤診。EPP肝臟受累、肝衰竭多出現(xiàn)在成人[7],鮮有早發(fā)型出現(xiàn)于兒童期[1],國內(nèi)關(guān)于EPP肝臟受累的報告更少[8]。筆者報告的4例患者均表現(xiàn)為急性/亞急性黃疸,臨床及實驗室檢查提示為肝內(nèi)膽汁淤積性黃疸(GGT、ALP升高為主,膽紅素以DBil升高為主,影像學(xué)缺乏肝外梗阻的證據(jù)),2例患者病情嚴(yán)重出現(xiàn)了脾功能亢進(jìn)(PLT降低)。
肝內(nèi)原卟啉沉積是EPP患者發(fā)生肝病的主要原因,因此肝組織中出現(xiàn)色素沉積物(原卟啉)是診斷EPP肝臟受累的直接證據(jù)。肝組織HE染色光鏡下可見肝細(xì)胞內(nèi)、肝細(xì)胞間微膽管內(nèi)、肝竇內(nèi)和巨噬細(xì)胞胞漿內(nèi)棕黃色顆粒樣沉積物沉積,這些沉積物實為原卟啉在肝臟內(nèi)的沉積。原卟啉具有結(jié)晶性質(zhì),因此在偏光顯微鏡下具有雙折光特性,呈現(xiàn)為“Maltese”十字樣,并隨著偏光顯微鏡的偏光濾光片角度的改變,“Maltese”十字的形狀及數(shù)目也發(fā)生改變。分光鏡研究表明這些色素的發(fā)射光譜與原卟啉標(biāo)準(zhǔn)液的發(fā)射光譜是一致的。4例患者均做了肝臟活組織檢查,偏光顯微鏡下也均證實了原卟啉在肝臟的淤積,從組織病理學(xué)角度提供了EPP肝臟受累的確鑿證據(jù)。因此對于鑒別肝內(nèi)膽汁淤積性肝病,除外病毒感染、藥物性等病因,還應(yīng)考慮到卟啉病等代謝性疾病,肝臟活組織檢查結(jié)合偏光顯微鏡有助于卟啉病的診斷。
EPP肝臟受累的治療方法有限,有研究認(rèn)為考來烯胺可阻斷原卟啉的腸肝循環(huán),促進(jìn)原卟啉從糞便中排出;此外熊去氧膽酸也有類似作用,可用于治療EPP。有部分研究者[9]采用反復(fù)輸血提供血紅素、血漿置換清除過多的原卟啉來治療EPP合并急性膽汁淤積。當(dāng)EPP合并肝功能衰竭,可以考慮肝移植[10],但是肝移植不能改變患者的基因缺陷,移植后仍可出現(xiàn)復(fù)發(fā),卟啉再次沉積于肝臟。有報道[11]認(rèn)為EPP合并肝功能衰竭患者可考慮肝移植后骨髓移植。
EPP是常染色體顯性遺傳,為不完全外顯,F(xiàn)ECH基因突變可以導(dǎo)致FECH活性下降。在顯性模式中,患者FECH活性為正常的10%~30%,而攜帶者FECH活性為正常的50%。國外關(guān)于FECH基因突變的報道相對較多,大多數(shù)突變是家族特異性的,尚未發(fā)現(xiàn)熱點(diǎn)突變。國內(nèi)這方面的報道很少,在一個EPP家系報道了剪接突變位點(diǎn)IVS3+1G→A[12]。本組4例患者均發(fā)現(xiàn)了FECH基因不同位點(diǎn)突變,這些突變在中國人群EPP患者中尚無明確報道,其致病性有待進(jìn)一步研究探討,但結(jié)合患者臨床表現(xiàn),支持EPP診斷。而且3例患者部分家系篩查也檢測到與患者相同的FECH突變位點(diǎn),其中患者1的哥哥亦是卟啉病患者。
因此,當(dāng)臨床上遇到年幼起病的日曬部位痛性紅斑、水腫、結(jié)痂,應(yīng)警惕EPP,皮膚活組織檢查、紅細(xì)胞內(nèi)原卟啉、FECH基因突變有助于診斷,并建議予以避光及β胡蘿卜素;這部分患者需要定期監(jiān)測肝功能和腹部超聲,以期早期發(fā)現(xiàn)EPP肝臟受累,肝臟組織學(xué)偏光顯微鏡下“Maltese”十字提示卟啉肝臟沉積。皮膚科、消化科、肝病科醫(yī)生應(yīng)該提高對EPP的認(rèn)識,以避免誤診、漏診。
[1] KHALIL MJ, FARAHMAD F, HIRBOD-MOBARAKEH A, et al. Erythropoietic protoporphyria and early onset of cholestasis[J]. Turk J Pediatr, 2012, 54(6): 645-650.
[2] WANG T, LIU YH, FANG K. A case of erythropoietic photoporphyria[J]. J Clin Dermatol, 2011, 40(6): 361-362. (in Chinese) 王濤, 劉躍華, 方凱. 紅細(xì)胞生成原性卟啉病1例[J]. 臨床皮膚科雜志, 2011, 40(6): 361-362.
[3] CONG L, WANG WL, XIA ZK, et al. A case of erythropoietic photoporphyria and literature review[J]. J Practical Dermatol, 2012, 5(3): 129-131. (in Chinese) 叢林, 王文嶺, 夏志寬, 等. 紅細(xì)胞生成性原卟啉病一例并文獻(xiàn)復(fù)習(xí)[J]. 實用皮膚病學(xué)雜志, 2012, 5(3): 129-131.
[4] LI Q, GAO TW. A case of erythropoietic photoporphyria[J]. Chin J Dermatovenereol, 2003, 17(3): 209-210. (in Chinese) 李強(qiáng), 高天文. 紅細(xì)胞生成性原卟啉病1例[J]. 中國皮膚性病學(xué)雜志, 2003, 17(3): 209-210.
[5] YANG S, YANG CJ, LIN D, et al. Two cases of erythropoietic photoporphyria[J]. Chin J Lepr Skin Dis, 2003, 19(2): 159-160. (in Chinese) 楊森, 楊春俊, 林達(dá), 等. 紅細(xì)胞生成性原卟啉病2例[J]. 中國麻風(fēng)皮膚病雜志, 2003, 19(2): 159-160.
[6] WU M, YU CP, LU XM. Two cases of erythropoietic photoporphyria[J]. Chin J Lepr Skin Dis, 2007, 23(5): 427-428. (in Chinese) 吳梅, 于長平, 盧憲梅. 紅細(xì)胞生成性原卟啉病2例[J]. 中國麻風(fēng)皮膚病雜志, 2007, 23(5): 427-428.
[7] REISENAUER AK, SOON SL , LEE KK, et al. Erythropoietic protoporphyria presenting with liver failure in adulthood[J]. Dermatology, 2005, 210(1): 72-73.
[8] CHEN JF, ZHANG P. Microstructure study of liver tissue in a patient of erythropoietic protoporphyria[J]. Acta Univ Med Nanjing, 2001, 21(6): 552-553. (in Chinese) 陳錦飛, 張平. 紅細(xì)胞生成性原卟啉病1例患者肝組織的顯微結(jié)構(gòu)研究[J]. 南京醫(yī)科大學(xué)學(xué)報, 2001, 21(6): 552-553.
[9] HONDA Y, KAWAKAMI Y, KAN H, et al. A second attack of cholestasis associated with erythropoietic protoporphyria was successfully treated by plasma exchange and blood transfusion[J]. Clin J Gastroenterol, 2014, 7(4): 333-337.
[10] LOCK G, HOLSTEGE A, MUELLER AR, et al. Liver failure in erythropoietic protoporphyria associated with choledocholithiasis and severe post-transplantation polyneuropathy[J]. Liver, 1996, 16(3): 211-217.
[11] CHEUNG CY, TAM S, LAM CW, et al. Allogeneic haematopoietic stem cell transplantation for erythropoietic protoporphyria: a cautionary note[J]. Blood cells Mol Dis, 2015, 54(3): 266-267.
[12] MA JH, XIAO SX, AN JG, et al. Mutation analysis of ferrochelatase gene in a pedigree with erythropoietic protoporphyria[J]. Chin J Dermatol, 2010, 43(2): 85-87. (in Chinese) 馬俊紅, 肖生祥, 安金剛, 等. 紅細(xì)胞生成性原卟啉病一家系亞鐵螯合酶基因突變檢測[J]. 中華皮膚病雜志, 2010, 43(2): 85-87.
引證本文:LI XQ, SHI J, LAI YM, et al. Erythropoietic protoporphyria with jaundice as the main manifestation: a clinical, pathological, and genetic analysis of 4 cases[J]. J Clin Hepatol, 2017, 33(7): 1332-1335. (in Chinese) 李曉青, 師杰, 賴雅敏, 等. 4例以黃疸為主要表現(xiàn)的紅細(xì)胞生成性原卟啉病臨床、病理及遺傳學(xué)分析[J]. 臨床肝膽病雜志, 2017, 33(7): 1332-1335.
(本文編輯:劉曉紅)
Erythropoietic protoporphyria with jaundice as the main manifestation: a clinical, pathological, and genetic analysis of 4 cases
LIXiaoqing,SHIJie,LAIYamin,etal.
(DepartmentofGastroenterology,PekingUnionMedicalCollegeHospital,PekingUnionMedicalCollege,ChineseAcademyofMedicalSciences,Beijing100730,China)
Objective To investigate the clinical, pathological, and genetic features of erythropoietic protoporphyria, and to enhance the knowledge of liver involvement in this disease. Methods A retrospective analysis was performed for the clinical data of 4 patients with erythropoietic protoporphyria with jaundice as the main manifestation who were hospitalized in Peking Union Medical College Hospital from July 2011 to September 2014, including clinical features, liver pathology, and gene mutations. Results All the children had an acute/subacute onset, with intrahepatic cholestatic jaundice as the main clinical feature. The liver function test showed gamma-glutamyl transpeptidase 425-1152 U/L, alkaline phosphatase 196-356 U/L, total bilirubin 287-485 μmol/L, and direct bilirubin 216-394 μmol/L. All the patients experienced typical skin pain, erythema, and blisters after sunshine in their childhood. Further examinations showed free erythrocyte protoporphyrin 49.8-113.1 μg/gHb, liver pathological examination showed “Maltese” cross under a polarizing microscope, and mutations at different loci of FECH gene were detected. Conclusion For patients with intrahepatic cholestatic liver disease with typical painful erythema after sunshine, the possibility of erythropoietic protoporphyria should be considered. Skin/liver pathology, erythrocyte protoporphyrin, and FECH gene detection help with the diagnosis of this disease.
protoporphyria, erythropoietic; jaundice; dermatitis, photoallergic
10.3969/j.issn.1001-5256.2017.07.026
2017-01-06;
2017-02-09。
李曉青(1976-),女,主治醫(yī)師,博士,主要研究消化科常見及疑難病,尤其是功能性胃腸病。
R589.8
A
1001-5256(2017)07-1332-04
猜你喜歡
肝博士(2024年1期)2024-03-12 08:38:08
傳染病信息(2022年6期)2023-01-12 08:58:58
肝博士(2022年3期)2022-06-30 02:48:58
昆明醫(yī)科大學(xué)學(xué)報(2021年3期)2021-07-22 07:39:56
中外醫(yī)療(2016年15期)2016-12-01 04:25:40
中國衛(wèi)生標(biāo)準(zhǔn)管理(2015年1期)2016-01-14 03:41:20
海軍醫(yī)學(xué)雜志(2015年2期)2015-02-27 13:47:43
癌變·畸變·突變(2015年4期)2015-02-27 06:15:18
中國藥業(yè)(2014年12期)2014-06-06 02:17:26
中國藥業(yè)(2014年19期)2014-05-17 03:12:13
