二硫化鉬/石墨烯異質結的界面結合作用及其對帶邊電位影響的理論研究?
2017-08-12 03:21:44危陽馬新國2祝林賀華2黃楚云2
物理學報 2017年8期
危陽 馬新國2)? 祝林 賀華2) 黃楚云2)?
1)(湖北工業大學理學院,武漢430068)2)(湖北工業大學,太陽能高效利用湖北省協同創新中心,武漢430068)
二硫化鉬/石墨烯異質結的界面結合作用及其對帶邊電位影響的理論研究?
危陽1)馬新國1)2)?祝林1)賀華1)2)黃楚云1)2)?
1)(湖北工業大學理學院,武漢430068)2)(湖北工業大學,太陽能高效利用湖北省協同創新中心,武漢430068)
(2016年12月15日收到;2017年1月21日收到修改稿)
采用基于色散修正的平面波超軟贗勢方法研究了二硫化鉬/石墨烯異質結的界面結合作用及其對電荷分布和帶邊電位的影響.研究表明二硫化鉬與石墨烯之間可以形成范德瓦耳斯力結合的穩定堆疊結構.通過能帶結構計算,發現二硫化鉬與石墨烯的耦合導致二硫化鉬成為n型半導體,石墨烯轉變成小帶隙的p型體系.并通過電子密度差分圖證實了界面內二硫化鉬附近聚集負電荷,石墨烯附近聚集正電荷,界面內形成的內建電場可以抑制光生電子-空穴對的復合.石墨烯的引入可以調制二硫化鉬的能帶,使其導帶底上移至?0.31 eV,提高了光生電子還原能力,有利于光催化還原反應.
異質結光催化,二硫化鉬,能帶調制,第一性原理
1 引言
隨著工業進程的加快,環境污染日益嚴重,太陽能作為安全無污染的新型能源受到了世界各國的廣泛關注.盡管它的應用前景已被廣泛接受,但因其傳統核心材料較小的光譜響應范圍和較低的太陽光利用率使其在實際應用中面臨諸多挑戰.如傳統光催化材料二氧化鈦只能吸收波長小于420 nm的紫外光,約占太陽光能量的3%—5%.因此,對于新型光催化材料的尋找迫在眉睫.片狀二硫化鉬因具有穩定的類石墨層狀結構、帶隙可調和可見光吸收特性,在催化[1]、太陽能電池[2]、光開關[3]和超級電容器[4]等領域具有廣闊的應用前景.尤其是單片層二硫化鉬具有較大的比表面積和較多的邊緣非飽和鍵,提供了豐富的活性位點和吸附位點,使其在光催化領域具有極大的應用潛力[5?7].但是單組分二硫化鉬光催化劑的能效并不高,主要原因在于較低的載流子傳輸能力和較高的光生電子-空穴復合率.為了解決這個問題,往往在二硫化鉬的納米體系中引入貴金屬、半導體或者共軛聚合物等進行異質結復合,形成肖特基結或者pn結,促進光生電子-空穴的分離,如Ag/MoS2[8],Bi2S3/MoS2[9],Bi2MoO6/MoS2[10],n-rGO/MoS2[11],P3 HT/MoS2[12]等,從而提高二硫化鉬的光催化活性.石墨烯具有極大的比表面積、較高的功函數以及優異的導電性能[13],與其他光催化劑復合,能夠達到增加光催化劑的吸附位點和抑制電子-空穴復合的雙重作用,提升光催化反應效率.Fu和Wang[14]發現鐵酸鋅/石墨烯復合材料有比純鐵酸鋅更高的可見光催化活性,其機理在于石墨烯的引入促進了過氧化氫的生成.Yun等[15]發現釩酸鉍和石墨烯復合有效提高了釩酸鉍在可見光下催化降解污染物的能力,原因在于釩酸鉍受光激發產生的光生電子能持續注入石墨烯中,使得載流子分離,從而提高光生電子的生命周期.此外,Xu等[16]還發現氧化鋅/石墨烯復合材料有比純氧化鋅更高的紫外光催化活性,這得益于石墨烯較高的電子遷移率.當前石墨烯與二硫化鉬異質結復合的研究已經成為關注的熱點之一,主要歸因于這種復合可以協同發揮石墨烯高導電性能和二硫化鉬光催化活性位點豐富的優勢[17?19].實驗上往往采用水熱法或者原位生長法等[20?22]制備石墨烯/二硫化鉬異質結復合體系,探討界面微結構和表界面光催化反應過程,發現石墨烯作為助催化劑可以提高光催化材料的分散性和產生更多的催化活性點[23],使光量子效率及產氫速率大大提升.
事實上,石墨烯與二硫化鉬異質結復合體系的光催化性能及其應用方面取得了重要進展,光量子效率已經超過24%[20],但是大量的研究主要集中在合成工藝、催化活性及性能的表征上,而石墨烯與二硫化鉬的結構特殊性使其片層之間可能存在較弱的相互作用,很難通過實驗直接進行探測,兩者形成的肖特基結[24]或者pn結[21,25]屬性也沒有形成一致意見,因此有必要對其界面結合作用與電子結構之間的關聯性進一步深入研究.采用第一性原理的方法可以獲得實驗研究無法獲取的有用信息,從材料設計的角度分析微結構對光電性能的影響.最近有研究人員采用該方法研究了它們的界面結合及能帶結構[24,26],但是有關界面內電荷分布狀況以及對能級匹配程度影響的系統研究仍然沒有展開,所表現出較強的光吸收能力和較高的載流子遷移率的物理機理仍然不清.為此,我們建立了晶格匹配程度較高的二硫化鉬/石墨烯異質結界面模型,采用基于密度泛函理論的第一性原理方法研究了界面結合作用以及其對電荷分布和帶邊電位的影響,探討了異質結界面內光生載流子遷移的過程,為進一步改善二硫化鉬基光催化劑的光電性能提供理論指導.
2 計算方法和物理模型
采用基于密度泛函理論的平面波超軟贗勢方法[27]研究了二硫化鉬/石墨烯異質結的界面結合作用以及其對電荷分布和帶邊電位的影響.為了能準確描述層間的范德瓦耳斯力,分別在廣義梯度近似(generalized-gradient app roxiMation,GGA)的Perdew-Burke-Ernzerhof(PBE)方案中考慮了Tkatchenko-Scheffl er(TS)和Grimme色散修正[28,29],以及在局域密度近似(local-density approximation)的Ceperley-A lder-Perdew-Zunger方案中考慮了Ortmann-Bechstedt-SchMidt(OBS)色散修正[30].在描述離子實與價電子之間的相互作用時,選取的價電子組態分別為C:2s22p2,S:3s23p4,Mo:4s24p64d55s1.布里淵區k點網格均選取為5×5×4[31].采用Broyden-Fletcher-Goldfarb-Shanno算法對所有模型進行幾何結構優化,平面波截斷能設置為400 eV,自恰收斂精度設置為5.0×10?5eV/atom,原子間的力場收斂精度設置為1 eV/nm,最大應力設置為0.2GPa,最大位移不超過5×10?4nm.所有計算均由CASTEP軟件完成[32].
采用GGA-PBE方法對六方相二硫化鉬(空間群:P 63/mMc)的單胞進行幾何結構優化,晶格常數為a=b=0.318 nm,c=1.241 nm,同時對石墨烯的單胞也進行幾何優化,晶格常數為a=b=0.246 nm,它們與實驗值相比誤差均小于1%(二硫化鉬:a=b=0.316 nm,c=1.230 nm;石墨烯:a=b=0.246 nm)[33,34].具有層狀堆疊結構的六方相二硫化鉬容易剝離出單層二硫化鉬,它是中間一層為Mo原子,上下兩層均為S原子的類三明治結構.為了討論異質結的晶格匹配,建立了兩種單層二硫化鉬/石墨烯異質結的結構匹配模型.具體為:3×3周期結構的單層二硫化鉬超胞與4×4周期結構的石墨烯超胞匹配(圖1(a)),以及4×4周期結構的單層二硫化鉬超胞與5×5周期結構的石墨烯超胞匹配(圖1(b)).圖1中片層模型的真空層厚度均選為1.5 nm.
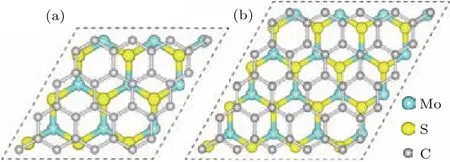
圖1 (網刊彩色)兩種單層二硫化鉬/石墨烯異質結匹配模型的頂視圖(a)單層二硫化鉬3×3超胞與石墨烯4×4超胞的匹配模型;(b)單層二硫化鉬4×4超胞與石墨烯5×5超胞的匹配模型Fig.1.(color on line)Top view s of two Match con figurations ofMonolayer MoS2/graphene heterostructu re:(a)Match con figuration between 3×3 lateral periodicity of Monolayer MoS2 sheet and 4×4 lateral periodicity of graphene;(b)Match con figu ration between 4×4 lateral periodicity ofMonolayer MoS2 sheet and 5×5 lateral periodicity of graphene.
3 結果與討論
3.1 結構穩定性
為了深入了解異質結的晶格匹配情況,基于圖1所示的物理模型,計算了二硫化鉬和石墨烯之間的晶格失配率.設定優化后異質結的平衡晶格常數為a′,單層二硫化鉬和石墨烯超胞的晶格常數分別為a1和a2,則晶格失配率可以定義為σ=(a2?a1)/a1.由此獲得圖1中兩種異質結匹配形式的晶格失配率分別為3.0%和?3.6%,可見這兩種異質結匹配模型均為完全共格(|σ|<5.0%).為了進一步確認異質結的結構穩定性,計算了其晶格失配能.這里晶格失配能定義為
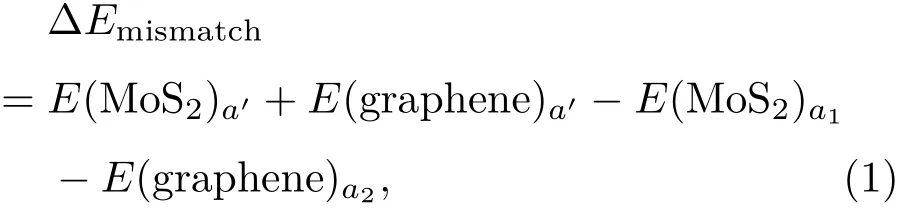
其中E(MoS2)a′和E(graphene)a′分別表示單層二硫化鉬和石墨烯的超胞晶格常數為a′時的總能量;E(MoS2)a1表示單層二硫化鉬晶格常數為a1時的總能量,E(graphene)a2表示石墨烯晶格常數為a2時的總能量.由此獲得兩種異質結匹配形式的晶格失配能分別為?0.542和?1.270 eV.事實上,晶格失配能的絕對值越低,異質結的結構越穩定.在隨后的研究中,僅選用失配率和失配能均較小的圖1(a)異質結模型展開研究.
為了確定二硫化鉬和石墨烯之間的層間作用類型,基于圖1(a)匹配結構的異質結模型,進一步構建了如圖2所示的四種層間對接模型.異質結的界面形成能可以表示為
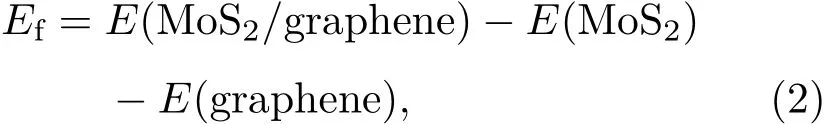
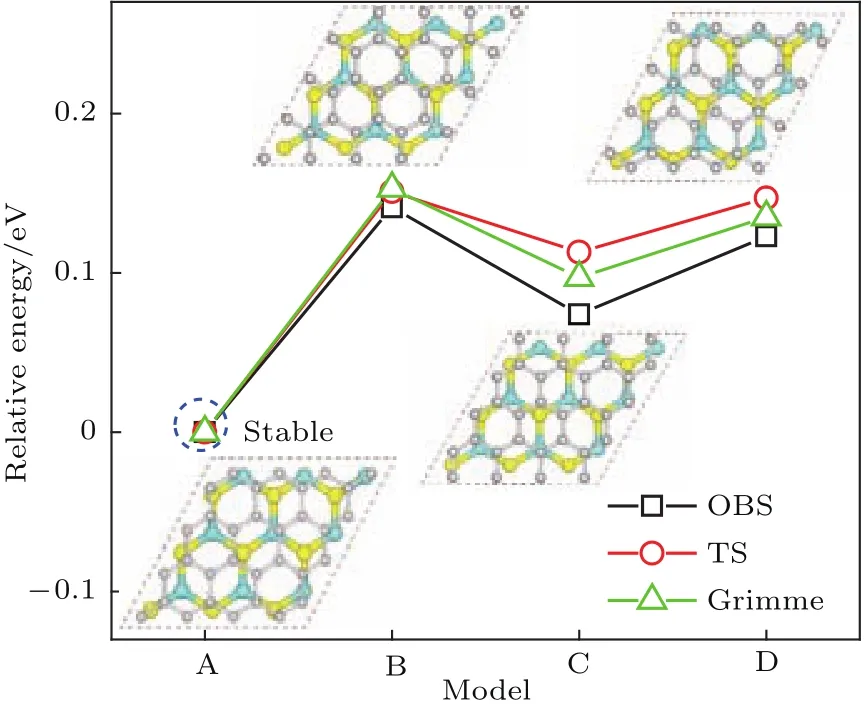
圖2 (網刊彩色)二硫化鉬/石墨烯異質結的四種層間疊層模型俯視圖以及采用三種色散修正方法獲得總能量,這里以疊層模型A的總能量為能量參考零點Fig.2.(color on line)Top view sand relative totalenergies using th ree dispersion correction Methods of fou r stacking patterns for MoS2/graphene heterostructu re.The total energy of stacking pattern A is set to zero.
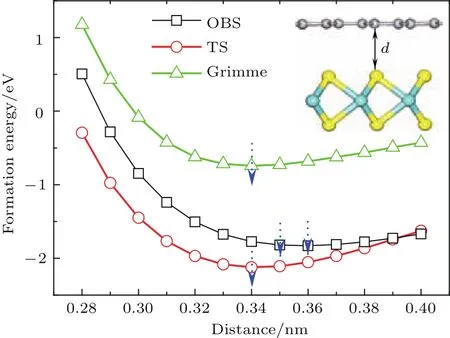
圖3 (網刊彩色)采用三種不同色散修正方法計算獲得的二硫化鉬/石墨烯異質結的層間距d與界面形成能之間的關系Fig.3.(color on line)The relation of interface forMation energy and layer d istance d of MoS2/graphene heterostructu re calcu lated using th ree dispersion correction Methods.
其中E(MoS2/graphene),E(MoS2)和E(graphene)分別表示弛豫二硫化鉬/石墨烯異質結、單層二硫化鉬和石墨烯的體系總能量.分別采用三種色散修正方法計算了異質結在不同層間距離d下的界面形成能,其關系曲線如圖3所示.可以看到TS和Grimme色散修正方法獲得最為穩定的層間距d約為0.34 nm,而OBS色散修正方法獲得最為穩定的層間距離約為0.35—0.36 nm,這與異質結模型經幾何優化后所得最優層間距約0.35 nm結果一致.實驗上得到的二硫化鉬/石墨烯異質結STEM圖譜顯示[35],石墨烯與二硫化鉬之間最穩定的間距約在(0.34±0.01)nm,這個結果與圖3結果相符.三種不同的色散修正方法計算所得異質結形成能隨層間距d變化的趨勢高度一致,僅能量上稍有差別,這與算法的選取有關.在層間距d約為0.28—0.34 nm時,隨著層間距增大,形成能逐步顯著降低,由正值變為負值,結構趨于穩定.此時層間相互作用主要表現為斥力,隨著層間距增大,斥力減弱,結構趨于穩定;在層間距d約為0.34—0.40 nm時,隨著層間距增大,形成能逐步緩慢增加,結構穩定性降低.此時層間相互作用主要表現為引力,隨著層間距增大,引力減弱,結構穩定性逐漸降低.這與文獻[36,37]結果一致.在四種層間疊層模型中,層間距與形成能的關系曲線趨勢均一致,所得最為穩定層間距離的結果也均相同.在此層間距離下,四種疊層模型的界面形成能均為負數,表明層間存在一定的結合作用,使其可以形成較為穩定的異質結.從兩個方面可以看出二硫化鉬和石墨烯之間存在范德瓦耳斯結合作用.一方面,四種疊層模型的界面形成能的大小非常接近,其差值不超過0.15 eV,表明層間的結合作用與它們層間對接方式并沒有太大的關系;另一方面,層間結合作用力均較小,例如圖2中模型A的單位面積界面形成能(結合能)僅為1.323 eV/nm2,在典型范德瓦耳斯結合能數值(1.3—2.1 eV/nm2)范圍內[38].實驗上測量了二硫化鉬/石墨烯異質結的光致發光(PL)譜[35],與二硫化鉬/二氧化硅的拉曼譜相比,二硫化鉬/石墨烯異質結的PL譜有微弱的上移(3 cm?1)和窄化(3 cm?1),由此認為兩者之間的相互作用力是范德瓦耳斯作用力.而我們所計算的界面結合能,同樣證明了二硫化鉬與石墨烯之間存在范德瓦耳斯結合作用.
3.2 能帶結構
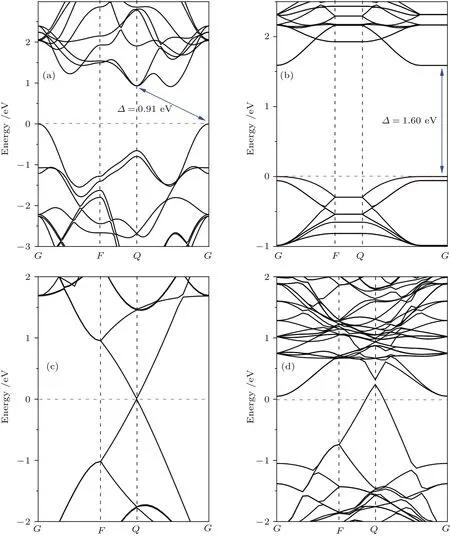
圖4 (網刊彩色)體相二硫化鉬(a)、單層二硫化鉬(b)、石墨烯(c)及二硫化鉬/石墨烯異質結(d)的能帶結構圖Fig.4.(color on line)E lectronic band structures of(a)MoS2 bu lk,(b)MoS2 Monolayer,(c)graphene and(d)MoS2/graphene heterostructure.The FerMi levels are set to zero and Marked by red dotted lines.
為了探究二硫化鉬/石墨烯異質結的界面結合作用對界面內電荷分布的影響,這里以圖2中模型A為例,計算了異質結的能帶結構和態密度.圖4為體相二硫化鉬、單層二硫化鉬、石墨烯以及二硫化鉬/石墨烯異質結的能帶結構.可以看出,體相二硫化鉬顯示了高對稱G點的價帶頂和高對稱Q點的導帶底之間0.91 eV的間接帶隙,而單層二硫化鉬顯示出高對稱G點位置的直接帶隙情形,其值為1.60 eV.Low等[39]實驗上測得體相和單層二硫化鉬的帶隙值分別為1.2和1.9 eV;Mak等[40]實驗上測得單層二硫化鉬的帶隙值為1.8 eV;Huang等[41]通過理論計算獲得單層二硫化鉬的帶隙值為1.67 eV.可以發現,我們的計算結果接近其他的理論計算結果,而略低于實驗值.由于密度泛函理論在計算帶隙值時普遍存在不可避免的誤差,GGA近似往往會高估晶格常數并低估帶隙值,但不影響能帶結構以及其他方面結果的分析.與純石墨烯的能帶結構比較,二硫化鉬/石墨烯異質結能帶結構中的費米能級發生了移動,使其位于石墨烯狄拉克點的下方,石墨烯導帶存在未填充能級,石墨烯形成了帶隙值不超過0.1 eV的p型體系.而費米能級位于二硫化鉬帶隙內的導帶底附近,使二硫化鉬顯示出典型的n型半導體特征.由圖4(b)—圖4(d)可以看出,石墨烯狄拉克點的位置高于單層二硫化鉬導帶底的位置,即兩者間存在電勢差.因此,當二硫化鉬與石墨烯形成異質結后,界面電荷將發生相對轉移:處于較高位置的石墨烯的電子將流入二硫化鉬表面,即二硫化鉬表面聚集負電荷,石墨烯表面聚集正電荷,這與計算所得電荷密度差分圖(圖5)顯示的結果一致.由于電荷的重新分布,導致石墨烯電勢增加,能帶下移;二硫化鉬電勢降低,能帶上移,這也被后面計算出的功函數所證實.
異質結的形成改變了二硫化鉬和石墨烯的電學屬性,這主要源于層間的結合作用導致的界面內電荷的重新分布.圖5(a)顯示了二硫化鉬/石墨烯異質結的電荷密度差分圖,其中紅色分布區域表示電子減少(正電荷聚集),藍色區域表示電子增加(負電荷聚集).可以看出,異質結界面的形成,導致石墨烯上的部分電子向二硫化鉬轉移,從而使二硫化鉬層聚集了較多的負電荷,石墨烯聚集了較多的正電荷,界面內形成了內建電場.二硫化鉬/石墨烯異質結界面的能帶結構和電荷分布原理如圖5(b)所示.Roy等[42]從實驗上研究了石墨烯/二硫化鉬異質結的光電流效應,當背柵電遠小于二硫化鉬的傳導閾值時,光生電子由二硫化鉬向石墨烯轉移,光生空穴由石墨烯向二硫化鉬轉移,這個結果與我們的研究結論一致.
為了更好地了解二硫化鉬/石墨烯異質結層間相互作用的微觀機理,分析了異質結的總態密度和分態密度,如圖6所示.可以看出,在費米能級附近異質結的價帶頂主要由Mo 4d和C 2p軌道組成,而導帶主要由Mo 4d和S 3p軌道組成.因此,在太陽光照下光生電子可以從價帶中Mo 4d軌道躍遷至導帶中Mo 4d軌道.從圖6(c)中可以看出導帶中Mo 4d和S 3p之間存在能級重疊,顯示了他們間存在較強的軌道雜化作用,使處于激發狀態下的電子很容易從Mo 4d軌道轉移至S 3p軌道,激發的電子趨向集中于界面間的S原子層.結合圖5可知,在界面內建電場的作用下,S原子附近的光生電子有向石墨烯轉移的趨勢,從而有利于光生電子-空穴對的有效分離.
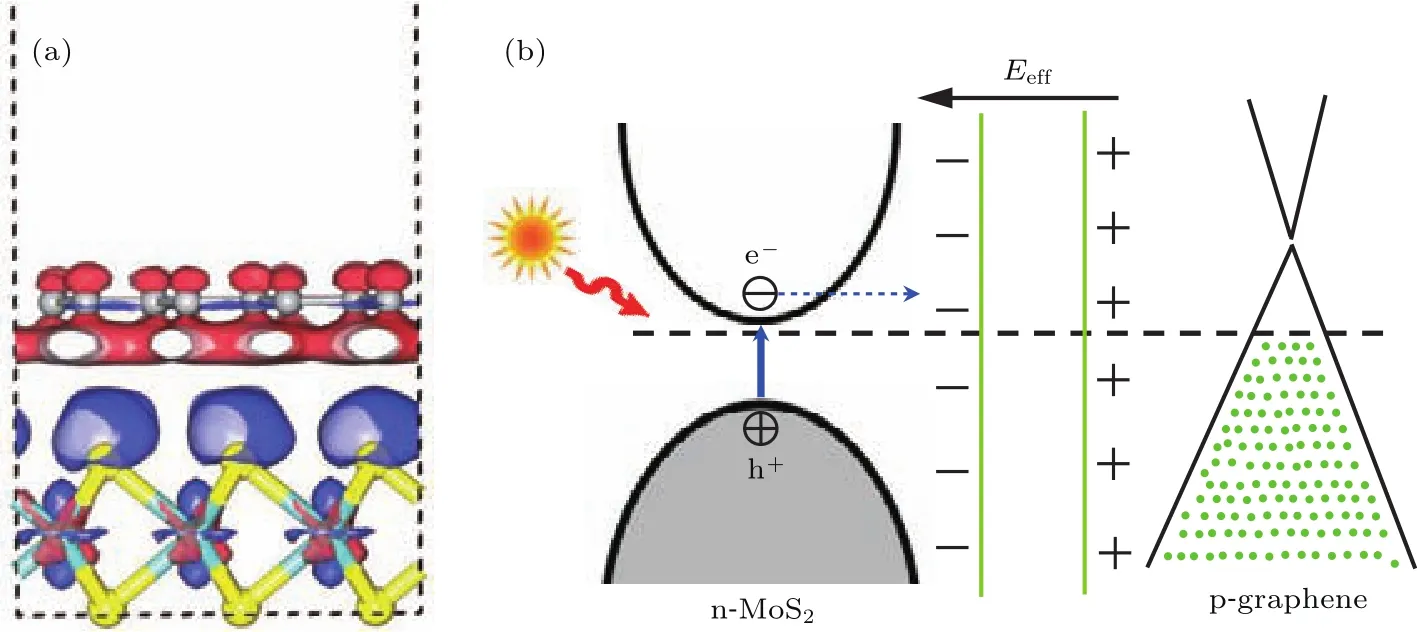
圖5 (網刊彩色)二硫化鉬/石墨烯異質結的三維電子密度差分圖以及界面電荷重新分布原理Fig.5.(color on line)Three-d iMensional charge density d iff erence p lots and p rincip le of the charge red istribution of MoS2/graphene heterostructure.
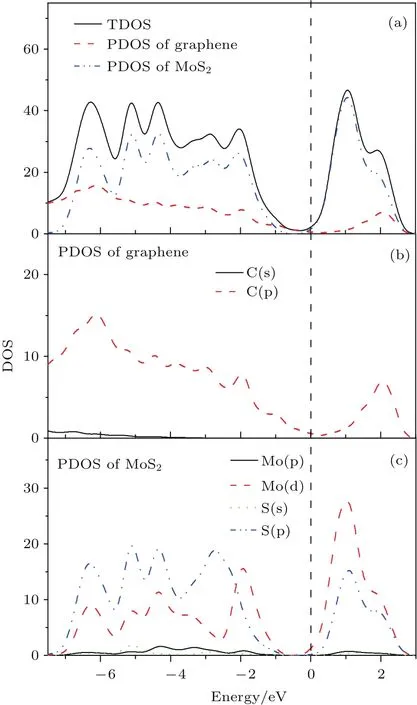
圖6 (網刊彩色)二硫化鉬/石墨烯異質結的總態密度以及相應的分態密度圖Fig.6.(color on line)Calcu lated total density of states(TDOS)and corresponding partial density of states(PDOS)of MoS2/graphene heterostructu re.
3.3 能級調制
異質結費米能級的電位是由異質結內兩體系的電子電離能共同決定.可以通過計算他們的功函數,獲得異質結費米能級的電位.功函數可以表示為
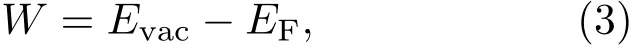
其中Evac是界面附近真空層靜態電子的能量,EF是費米能級的能量,用于決定基態電子結構的計算.圖7(a)—圖7(c)中藍色虛線代表理論計算所得真空層的位置,即Evac;紅色虛線代表理論計算所得費米能級的位置,即EF.這里計算出單層二硫化鉬、石墨烯和異質結的功函數分別為4.89,4.43和4.55 eV,單層二硫化鉬和石墨烯的功函數計算結果與文獻[43,44]基本一致,其值分別為4.92和4.50 eV.實驗上采用了次級電子帶邊測量的方法測得二硫化鉬/石墨烯異質結功函數[35],得到其實驗值約為4.40 eV.這與我們計算所得異質結功函數的數值4.55 eV大體一致,誤差僅為3%.存在的微小誤差一般是由于計算方法和精度選取不同而導致的,在選取相同方法和精度的情況下并不會影響計算的整體結果.由于石墨烯的功函數低于二硫化鉬,當兩者接觸時,部分電子將從石墨烯層轉移至二硫化鉬層,使二硫化鉬層形成電子為多子的n型半導體,二硫化鉬的費米能級上移0.34 eV,石墨烯的費米能級下移0.12 eV.
由于半導體光催化劑的價帶頂和導帶底電位直接決定了其光催化氧化還原能力,因此較準確的計算半導體的帶邊位置十分重要.半導體帶邊的電位可以采用平均電負性法進行估算,具體可以表示為[45,46]
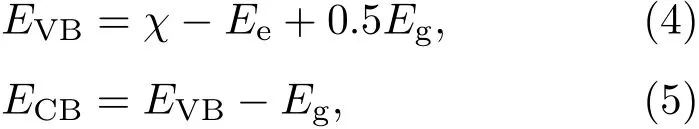
其中χ表示多元化合物半導體的絕對平均電負性;Ee是氫能級上自由電子的能量,其值為4.5 eV;Eg是半導體帶隙.由此得到二硫化鉬導帶底和價帶頂的電位分別為?0.13和1.77 eV,如圖7(d)所示.當兩者接觸形成異質結后,二硫化鉬的邊帶位置會隨著兩者費米能級的持平而改變.計算出的二硫化鉬/石墨烯異質結的功函數4.55 eV介于二硫化鉬和石墨烯兩者功函數之間,這是界面電荷轉移所致.
根據熱力學平衡條件,在電場力和擴散力的作用下兩者的費米能級將實現平衡.盡管二硫化鉬的費米能級上移0.34 eV,但同時異質結內的雜化作用使二硫化鉬的帶隙減少0.32 eV,致使導帶底產生約0.16 eV的下移,因而總體上二硫化鉬的導帶底僅上移約0.18 eV,此時導帶底的電位為?0.31 eV.進一步可以推斷價帶頂的電位約為1.27 eV.由此可見,石墨烯的引入使二硫化鉬的導帶電位更負,光生電子的還原能力更強.實驗表明二硫化鉬/石墨烯異質結具有比單獨的二硫化鉬或石墨烯更高的光催化性能[18,20,21,34].二硫化鉬與石墨烯接觸后,它的導帶底電位由?0.13 eV變為?0.31 eV,比氫氣(0 eV)和氧氣(?0.28 eV)的電位更負,能將氫離子還原為氫氣,同時部分光生電子和氧分子反應能產生具有強氧化能力的超氧自由基降解部分有機污染物.
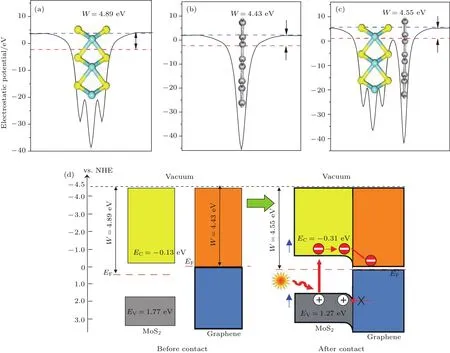
圖7 (網刊彩色)單層二硫化鉬(a)、石墨烯(b)及二硫化鉬/石墨烯異質結(c)的功函數圖,以及二硫化鉬和石墨烯接觸前后的邊帶位置圖(d)Fig.7.(color on line)Calcu lated work functions for(a)Monolayer MoS2,(b)graphene,(c)MoS2/graphene heterostructure;(d)the band edge positions before and after contact of MoS2 and graphene.The green and b lue lines denote FerMi level and the vacuuMenergy level,respectively.
4 結論
采用平面波超軟贗勢方法研究了二硫化鉬/石墨烯異質結的界面結合作用和電學性質.計算出的界面形成能顯示出二硫化鉬與石墨烯之間可以形成范德瓦耳斯力相結合的堆疊結構.通過能帶結構的計算,可以發現石墨烯的耦合改變了二硫化鉬的半導體性質,使其形成顯著的n型半導體.電子密度差分圖進一步表明了在界面內形成了由石墨烯指向二硫化鉬的內建電場.光照下光生電子通過Mo原子的d-d軌道躍遷至導帶,由于導帶中Mo 4d和S 3p之間存在較強的軌道雜化作用,使處于激發狀態下的電子從MoS2層內的Mo原子轉移至界面中的S原子,促進了電子-空穴的分離.通過功函數計算,發現石墨烯的引入可以調制二硫化鉬的能帶,使其導帶底上移,提高了導帶內光生電子的還原能力,有利于光催化還原反應.
[1]Li Y,W ang H,X ie L,Liang Y,Hong G,Dai H 2011 J.Am.Chem.Soc.133 7296
[2]Bernardi M,PalumMo M,G rossMan J C 2013 Nano Lett.13 3664
[3]B ritnell L,Ribeiro R M,EckMann A,Jalil R,Belle B D,Mishchenko A,K iMY J,Gorbachev R V,Georgiou T,Morozov S V,G rigorenko A N,GeiMA K,Casiraghi C,Neto A H C,Novoselov K S 2013 Science 340 1311
[4]Patil S,Harle A,Sathaye S,Patil K 2014 Cryst.Eng.Comm.16 10845
[5]N?rskov J K,B ligaard T,Logadottir A,K itchin J R,Chen JG,Pandelov S,StimMing U 2005 J.Electrochem.Soc.152 J23
[6]K arunadasa H I,Montalvo E,Sun Y J,Ma jda M,Long J R,Chang C J 2012 Science 335 698
[7]Garrett B R,Polen SM,C lick K A,He MF,Huang Z J,Hadad C M,Wu Y Y 2016 Inorg.Chem.55 3960
[8]Cheah A J,Chiu W S,K hiew P S,Naka jiMa H,Saisopa T,Songsiriritthigu l P,RadiMan S,HaMid MA A 2015 Catal.Sci.Technol.5 4133
[9]W eng B,Zhang X,Zhang N,Tang Z R,Xu Y J 2015 LangMuir 31 4314
[10]Chen Y J,T ian G H,Shi Y H,X iao Y T,Fu H G 2015 Appl.Catal.B:Environ.164 40
[11]Meng F,Li J,Cushing S K,ZhiM,Wu N 2013 J.Am.Chem.Soc.135 10286
[12]Zhao M,Chang MJ,Wang Q,Zhu Z T,Zhai X P,Zirak M,Mosh fegh A Z,Song Y L,Zhang H L 2015 Chem.ComMun.51 12262
[13]Liu Z F,Liu Q,Huang Y,Ma Y F,Y in S G,Zhang X Y,Sun W,Chen Y S 2008 Adv.Mater.20 3924
[14]Fu Y S,W ang X 2011 Ind.Eng.Chem.Res.50 7210
[15]Yun H N,Iwase A,Kudo A,AMalR 2010 J.Phys.Chem.Lett.1 2607
[16]Xu T G,Zhang L W,Cheng H Y,Zhu Y F 2011 Appl.Catal.B:Environ.101 382
[17]Li H L,Yu K,Li C,Tang Z,Guo B J,Lei X,Fu H,Zhu Z Q 2015 Sci.Rep.5 18730
[18]Chang K,Mei ZW,W ang T,K ang Q,Ouyang S X,Ye J H 2014 ACS Nano 8 7078
[19]K uMar N A,Dar MA,Gu l R,Baek J B 2015 Mater.Today 18 286
[20]Min S X,Lu G X 2012 J.Phys.Chem.C 116 25415
[21]Carraro F,Calvillo L,Cattelan M,Favaro M,Righetto M,NappiniS,Pí?I,Celorrio V,Fermín D J,Martucci A,Agnoli S,GranozziG 2015 ACS Appl.Mater.Interfaces 7 25685
[22]Deng Z H,Li L,DingW,Xiong K,Wei Z D 2015 Chem.ComMun.51 1893
[23]JaraMillo T F,J?rgensen K P,Bonde J,Nielsen J H,Horch S,Chorkendorff IB 2007 Science 317 100
[24]Jin C J,Rasmussen F A,Thygesen K S 2016 J.Phys.Chem.C 120 1352
[25]Aziza Z B,Henck H,Felice D D,Pierucci D,Chaste J,Naylor C H,Balan A,Dappe Y J,Johnson A T C,Ouerghi A 2016 Carbon 110 396
[26]Ebnonnasir A,Narayanan B,KodaMbaka S,Ciobanu C V 2014 Appl.Phys.Lett.105 031603
[27]Vanderbilt D 1990 Phys.Rev.B 41 7892
[28]Tkatchenko A,Scheffl er M2009 Phys.Rev.Lett.102 073005
[29]G rimMe S 2006 J.CoMput.Chem.27 1787
[30]O rtMann F,Bechsted t F,SchMid t W G 2006 Phys.Rev.B 73 205101
[31]Monkhorst H J,Pack J D 1976 Phys.Rev.B 13 5188
[32]Segall MD,Lindan P J D,Probert MJ,Pickard C J,Hasnip P J,C lark S J,Payne MC 2002 J.Phys:Condens.Matter 14 2717
[33]W u MS,Xu B,Liu G,Ouyang C Y 2012 Acta Phys.Sin.61 227102(in Chinese)[吳木生,徐波,劉剛,歐陽楚英2012物理學報61 227102]
[34]Jiang JW 2015 Front.Phys.10 287
[35]Pierucci D,Henck H,Avila J,Balan A,Nay lor C H,Patriarche G,Dappe Y J,Silly MG,Sirotti F,Johnson A T,Asensio MC,Ouerqhi A 2016 Nano Lett.16 4054
[36]Zhu J D,Zhang J C,Hao Y 2016 Jpn.J.App l.Phys.55 080306
[37]Ma Y D,Dai Y,Guo M,Niu C W,Huang B B 2011 Nanosca le 3 3883
[38]Liu J J 2015 J.Phys.Chem.C 119 28417
[39]Low J X,Cao SW,Yu JG,W ageh S 2014 Chem.ComMun.50 10768
[40]Mak K F,Lee C,Hone J,Shan J,Heinz T F 2010 Phys.Rev.Lett.105 136805
[41]Huang Z Y,He C Y,Qi X,Yang H,Liu W L,W ei X L,Peng X Y,Zhong J X 2014 J.Phys.D:Appl.Phys.47 75301
[42]Roy K,PadManabhan M,GoswaMi S,Sai T P,RaMalingaMG,Gaghavan S,Ghosh A 2013 Nat.Nano technol.8 826
[43]Liu B,Wu L J,Zhao Y Q,Wang L Z,Cai MQ 2016 RSC Adv.6 60271
[44]K iMJ H,Hwang JH,Suh J,Tongay S,Kwon S,Hwang C C,W u J Q,Park J Y 2013 App l.Phys.Lett.103 171604
[45]Xu Y,Schoonen MA A 2000 Am.Mineral.85 543
[46]Ma X G,Lu B,Li D,Shi R,Pan C S,Zhu Y F 2011 J.Phys.Chem.C 115 16963
(Received 15 December 2016;revised Manuscrip t received 21 January 2017)
PACS:71.15.Mb,71.20.–b,79.60.JvDOI:10.7498/aps.66.087101
Interfacial cohesive interaction and band Modu lation of tw o-d iMensional MoS2/graphene heterostructu re?
Wei Yang1)Ma Xin-Guo1)2)?Zhu Lin1)He Hua1)2)Huang Chu-Yun1)2)?
1)(School of Science,Hubei University of Technology,W uhan 430068,China)2)(Hubei Collaborative Innovation Center for High-Effi ciency Utilization of Solar Energy,Hubei University of Technology,W uhan 430068,China)
To iMp rove the effi ciency of water-sp litting,a key way is to select suitable seMiconductor or design seMiconductor based heterostructure to enhance charge separation of photogenerated h+-e?pairs.It is possible for a two-diMensional(2D)heterostructure to show More effi cient charge separation and transfer in a short transport tiMe and distance.AMong numerous heteromaterials,the 2D layered MoS2has become a very valuab lematerial in photocatalysis-driven field due to the appropriate electronic structure,peculiar thermal and cheMical stability,and low-cost preparation.To coup le w ith MoS2,layered graphene w ill be an ideal candidate due to extreMely high carrier Mobility,large surface area,and good latticeMatch w ith MoS2.A t p resent,a lot of researches focus on the synthesis and Modification of MoS2/graphene heterostructure.However,it is hard to detect directly the weak interaction between MoS2and graphene through the experiment.Here,an eff ective structural coup ling approach is described tomodify the photoelectrocheMical propertiesof MoS2sheet by using the stacking interaction w ith graphene,and the corresponding eff ects of interface cohesive interaction on the charge redistribution and the band edge of MoS2/graphene heterostructure are investigated by using the p lanewave ultrasoft pseudopotentials in detail.Three dispersion corrections take into account the weak interactions between MoS2and graphene,resulting in an equilibriuMlayer distance d of about 0.34 nMfor theMoS2/graphene heterostructure.The resu lts indicate that the latticeMismatch between monolayer MoS2and graphene is low in contact and a van der Waals interaction forMs in interface.Further,it is identified by analyzing the energy band structures and the threediMensional charge density diff erence that in the MoS2layer in interface there appears an obvious electron accumulation,which presents a new n-type seMiconductor for MoS2and a p-type graphene w ith a sMall band gap(<0.1 eV).In addition,Mo 4d electrons in the upper valence band can be excited to the conduction band under irradiation.And the orbital hybridization between Mo 4d and S 3p w ill cause photogenerated electrons to transfer easily froMthe internalMo atoMs to the external S atoMs.The build-in internal electric field froMgraphene to MoS2w ill facilitate the transfer and separation of photogenerated charge carriers after equilibriuMof the MoS2/graphene interface.It is identified that the hybridization between the two coMponents induces a decrease of band gap and then an increase of optical absorption of MoS2in visib le-light region.It is noted that their energy levels are ad justed w ith the shift of their FerMi levels based on our calculated work function.The results show that the FerMi level ofMonolayer MoS2is located under the conduction band and More positive than that of graphene.A fter the equilibriuMof the MoS2/graphene interface,the FerMi levelshifts toward the negative direction for MoS2and the positive direction for graphene,respectively,until they are equal.A t this tiMe,the conduction band and valence band of MoS2are pu lled to the negative direction a little,and then forMa slightly upward band bending close to the interface between MoS2and graphene.Combining the decrease of the band gap of MoS2in heterostructure,the potential of the conduction band MinimuMof MoS2in heterostructure w ill increase to?0.31 eV,which enhances its reduction capacity.A detailed understanding of theMicrocosMicMechanisMs of interface interaction and charge transfer in this systeMcan be help ful in fabricating 2D heterostructure photocatalysts.
heterostructure photocatalysis,MoS2,band modulation,fi rst-principles
10.7498/aps.66.087101
?國家自然科學基金(批準號:51472081)、湖北工業大學高層次人才啟動基金(批準號:GCRC13014)、綠色工業引領計劃(批準號:YXQN 2016005)和湖北省協同創新中心開放基金(批準號:HBSKFZD 2015004)資助的課題.
?通信作者.E-Mail:Maxg2013@sohu.com
?通信作者.E-Mail:chuyunh@163.com
?2017中國物理學會C h inese P hysica l Society
http://w u lixb.iphy.ac.cn
*Pro ject supported by the National Natural Science Foundation of China(G rant No.51472081),the Foundation of Hubei University of Technology for High-Level Talents,China(G rant No.GCRC 13014),the Leading P lan of G reen Industry,China(G rant No.YXQN 2016005),and the DevelopMent Founds of Hubei Collaborative Innovation Center,China(G rant No.HBSKFZD 2015004).
?Corresponding author.E-Mail:Maxg2013@sohu.com
?Corresponding au thor.E-Mail:chuyunh@163.com
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
當代陜西(2020年13期)2020-08-24 08:22:02
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
制造技術與機床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
新聞傳播(2015年11期)2015-07-18 11:15:04
