丙硫菌唑的合成研究
2017-09-03 03:46:11馬藝超張蒙萌申國富湯保賀程繹南李洪連
現代農藥 2017年4期
馬藝超,張蒙萌,申國富,湯保賀,王 寬,程繹南,李洪連
(河南農業大學 植保學院,鄭州 450002)
◆研究與開發◆
丙硫菌唑的合成研究
馬藝超,張蒙萌,申國富,湯保賀,王 寬,程繹南*,李洪連
(河南農業大學 植保學院,鄭州 450002)
以鄰氯芐基氯、2-氯-1-(1-氯環丙基)乙酮和水合肼等為主要原料,通過格氏反應、肼的取代、環化和氧化等4步反應合成了丙硫菌唑。目標產物及主要中間體經NMR確認,合成總收率達53%,產品質量分數為95%。該工藝具有反應條件相對溫和,反應溶劑易于回收和產品質量好等特點,較適合工業化開發。
丙硫菌唑;合成;格氏反應;取代肼
丙硫菌 唑(prothioconazole) 分 子 式 為C14H15Cl2N3OS,相對分子質量344.26,化學名2-[2-(1-氯環丙基)-3-(2-氯苯基)-2-羥基丙基]-2H-1,2,4-三唑-3(4H)-硫酮,CAS登錄號[178928-70-6]。其為白色或淡黃色固體,熔點為138~139℃。丙硫菌唑是拜耳公司于2004年開發上市的新型三唑硫酮類殺菌劑[1]。其具有廣譜殺菌活性,通過抑制甾醇前體羊毛甾醇的脫甲基化作用,干擾甾醇的生物合成,從而發揮殺菌活性[2]。丙硫菌唑內吸性良好,由于作用機理獨特,其具有優異的保護、治療和鏟除活性,主要用于禾谷類作物,如小麥、大麥、水稻、花生、油菜及豆類等,防治眾多病害。
由于其合成步驟長,難度大,丙硫菌唑在國內的開發與推廣應用尚處于初期階段。本文借鑒國內、外關于丙硫菌唑合成的相關文獻報道,選擇了如圖1所示的合成工藝路線[3-6]。
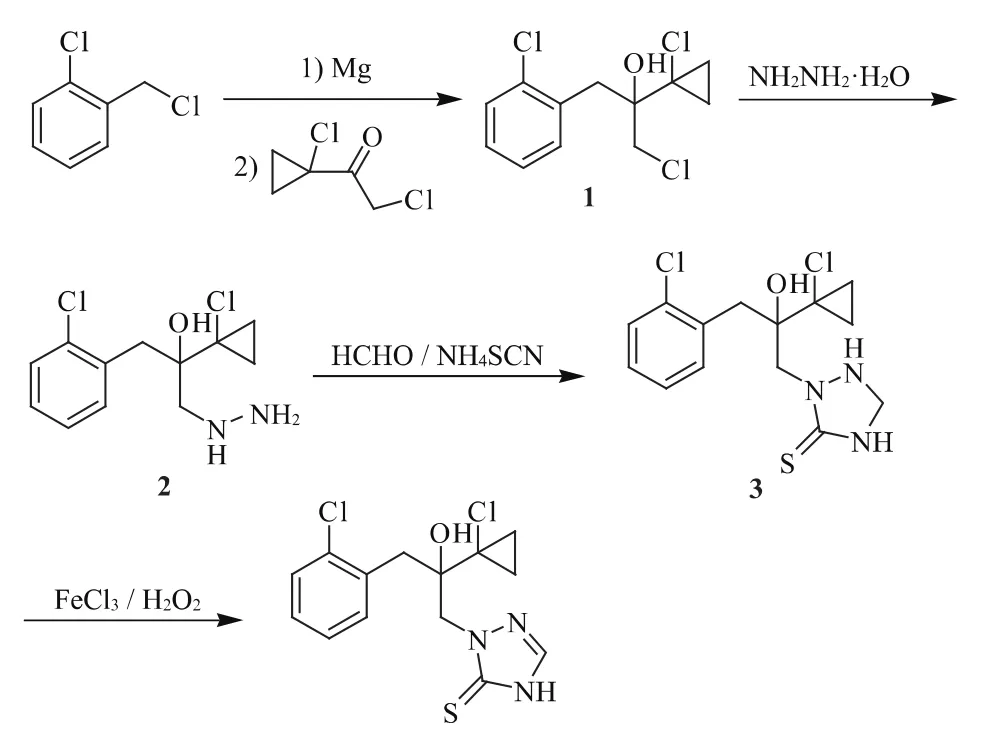
圖1 丙硫菌唑合成工藝
該合成工藝具有原料易得,反應條件溫和,溶劑能夠有效回收套用,中間體易于分離和純化等特點,總收率達到53%,具有較好的工業應用前景。
1 實驗部分
1.1 實驗儀器和試劑
2-氯-1-(1-氯環丙基)乙酮,購自薩恩化學技術(上海)有限公司;鄰氯芐基氯,購自鄭州阿爾法試劑公司。所有試劑未經純化直接使用。用青島海洋化工廠生產的硅膠GF254與羧甲基纖維素鈉水溶液(質量分數3.5‰)制備薄層色譜板。
實驗儀器:1H NMR及13C NMR用Bruker DPX-400型超導核磁共振儀測定,CDCl3或DMSO為溶劑,TMS為內標;GC211A氣相色譜儀;GC-MS 6800氣相色譜質譜聯用儀;Agilent1200液相色譜儀。
1.2 實驗方法
1.2.1 1-氯-2-(1-氯環丙基)-3-(2-氯苯基)-2-丙醇(中間體1)的合成
將5.3 g(0.22 mol)鎂屑投入到盛有50 mL無水叔丁基甲醚的反應瓶中,加入少量的碘和二溴乙烷,再加入1 g鄰氯芐基氯,加熱至回流。緩慢滴入溶有32.2 g(0.2 mol)鄰氯芐基氯的100 mL叔丁基甲醚溶液,鄰氯芐基氯的叔丁基甲醚溶液滴加完畢后,繼續回流反應2 h,得到灰色格氏試劑懸浮液。將得到的格氏試劑懸浮液降溫至10℃以下,緩慢加入含有24.5 g(0.16 mol)2-氯-1-(1-氯環丙基)乙酮的叔丁基甲醚溶液(50 mL),加畢將反應體系升至室溫繼續反應3 h。用飽和氯化銨水溶液淬滅反應,并分出有機層,用叔丁基甲醚萃取水相2次,合并有機相,有機相干燥濃縮后得黃色液體1-氯-2-(1-氯環丙基)-3-(2-氯苯基)-2-丙醇(中間體1)41 g,質量分數86%,收率79%。
1H NMR(400 MHz,CDCl3)δ:7.46-7.48(m,1H,Ar-H), 7.35-7.38(d,1H,Ar-H),7.19-7.21(m,2H,Ar-H),4.13-4.16(d, J=11.2 Hz,1H,CH-Cl),3.71-3.74 (d,J=11.6 Hz,1H,CH-Cl), 3.65-3.68(d,J=14.4 Hz,1H,Ar-CH),3.14-3.18(d,J=14 Hz, 1H,Ar-CH),2.41(s,1H,OH),0.84-1.25(m,4H,CH2CH2)。
1.2.2 2-(1-氯環丙基)-1-(2-氯苯基)-3-肼基-2-丙醇(中間體2)的合成
向裝有回流冷凝管的250 mL反應瓶中投入32.5 g(0.1 mol)1-氯-2-(1-氯環丙基)-3-(2-氯苯基)-2-丙醇(中間體1)和50 mL水,然后加入40 g(1.0 mol)80%水合肼,加熱回流反應10 h,當1-氯-2-(1-氯環丙基) -3-(2-氯苯基)-2-丙醇轉化完全后,反應液降至室溫,分層。有機相水洗2次,得到黏稠狀液體(中間體2)29.6 g,質量分數88%,收率95%。該中間體無需進一步純化,直接用于下步反應。
1.2.3 2-[2-(1-氯環丙基)-3-(2-氯苯基)-2-羥基丙基] -1,2,4-三唑烷-3-硫酮(中間體3)的合成
向盛有100 mL甲苯的250 mL反應瓶中,投入25 g(80 mmol)2-(1-氯環丙基)-1-(2-氯苯基)-3-肼基-2-丙醇,室溫攪拌下加入6.0 g(80 mmol)40%甲醛水溶液,0.5 h后,加入6.1 g(80 mmol)硫氰酸胺,繼續反應5 h,待原料轉化完畢后分層,有機相用水洗2次,干燥濃縮后得固體2-[2-(1-氯環丙基)-3-(2-氯苯基)-2-羥基丙基]-1,2,4-三唑烷-3-硫酮(中間體3)26 g,質量分數81%,收率76%。分析樣品由簿層色譜分離,熔點151~153℃。
1H NMR(400 MHz,CDCl3)δ:7.54-7.56(d,J=6.4 Hz,1H, Ar-H),7.35-7.36 (d,J=7.2 Hz,1H,Ar-H),7.19-7.21 (m,2H, Ar-H),6.43(s,1H,S=C-NH),5.10-5.15(t,J=10.8 Hz,J=10.4 Hz,NH),4.47-4.60(m,3H,OH and Ar-CH2),4.16(s,2H, NCH2N),3.61-3.64(d,J=14 Hz,1H,Ar-H),3.06-3.09(d,J=14 Hz,1H,Ar-H),0.86-1.25(m,4H,CH2CH2)。
13C NMR(100 MHz,CDCl3)δ:181.7(1C,C=S),135.1(1C, phenyl),134.2 (1C,phenyl),133.5 (1C,phenyl),129.3(1C, phenyl),128.1 (1C,phenyl),126.3 (1C,phenyl),76.9(1C, C-OH),61.6(1C,N-CH2-N),54.4(1C,Cl-C),46.2(1C,N-C), 38.2(1C,phenyl-C),11.2(1C,CH2),11.1(1C,CH2)。
1.2.4 丙硫菌唑的合成
將上步合成的20 g(50 mmol)2-[2-(1-氯環丙基) -3-(2-氯苯基)-2-羥基丙基]-1,2,4-三唑烷-3-硫酮投入盛有50 mL乙腈的250 mL反應瓶中,在室溫攪拌下加入30 g含有50 mmol三氯化鐵的水溶液,然后再加入5.7 g含有50 mmol雙氧水的水溶液,室溫繼續攪拌6 h,直到轉化完全。加入100 mL乙酸乙酯萃取,有機相用30 mL飽和亞硫酸鈉水溶液洗滌2次,干燥、濃縮,冷卻后析出淡黃色固體目標物16.8 g,質量分數95%,收率93%。分析樣品由簿層色譜分離,熔點137~138℃(文獻值138~139℃[7])。
1H NMR(400 MHz,CDCl3)δ:7.86(s,1H,N=CH-N),7.53-7.55(d,J=6.8 Hz,1H,Ar-H),7.36-7.38(d,J=7.2 Hz,1H, Ar-H),7.20-7.23(m,2H,Ar-H),4.77-4.81(d,J=14.4 Hz,1H, CH-N),4.47-4.51(d,J=14.8 Hz,1H,CH-N),4.24(s,1H,OH), 3.59-3.63(d,J=14 Hz,1H,Ar-CH),3.16-3.19(d,J=14 Hz,1H, Ar-CH),0.76-0.94(m,4H,CH2CH2)。
13C NMR(100 MHz,CDCl3)δ:165.6(1C,C=S),137.4 (1C,N-C=N),135.2(1C,phenyl),133.9(1C,phenyl),133.5(1C,phenyl),129.5(1C,phenyl),128.3(1C,phenyl),126.4 (1C,phenyl),77.2(1C,C-OH),53.9(1C,Cl-C),45.6(1C,N-C), 38.3(1C,phenyl-C),11.2(1C,CH2),11.1(1C,CH2)。
2 結果與討論
2.1 1-氯-2-(1-氯環丙基)-3-(2-氯苯基)-2-丙醇的合成討論
在1-氯-2-(1-氯環丙基)-3-(2-氯苯基)-2-丙醇的合成中,由于鄰氯芐基氯的芐基位具有較高的化學反應活性,以至于在格氏試劑的反應中時常伴有芐基位偶聯產物的生成,因此鄰氯芐基格氏試劑的合成是該反應的關鍵步驟。偶聯反應的發生存在2種可能:一種是所形成的格氏試劑在氧化劑的作用下發生自身偶聯[8];另一種是所形成的格氏試劑與另一分子鄰氯芐基氯反應而得。前者需要在無氧條件下操作,而后者需要控制格氏試劑的反應活性,如控制較低的反應溫度,選擇對格氏試劑具有穩定作用的反應溶劑等。為此,我們以碘(I2)為引發劑,在惰性氣體(N2)保護下,控制相對較低的反應溫度,來考察不同的反應溶劑對格氏試劑的形成難易及對偶聯反應的影響,反應溶劑包括乙醚、叔丁基甲醚(MTBE)、四氫呋喃(THF)、MTBE/甲苯和THF/甲苯。當以乙醚為溶劑時,反應在室溫下即可引發,并且反應的專一性強,反應的收率在95%以上,偶聯產物可以得到有效控制。然而,由于乙醚具有較低的沸點和爆炸危險性,不適用于規模生產。以叔丁基甲醚為溶劑時,反應室溫下難以引發,但當使用I2和二溴乙烷作為復合引發劑時,在回流條件下反應可以引發,反應收率達到79%,偶聯產物可以控制在10%以內[9]。雖然以叔丁基甲醚為溶劑的反應收率不及乙醚,但其相對穩定,可以滿足規模生產的要求。以THF為溶劑,反應室溫下能夠引發,但幾乎都生成了偶聯產物。MTBE/甲苯和THF/甲苯2種混合溶劑體系分別在反應引發和偶聯產物的控制方面表現不夠理想。目前比較適應規模生產的反應溶劑為MTBE。
2.2 2-[2-(1-氯環丙基)-3-(2-氯苯基)-2-羥基丙基]-1,2,4-三唑烷-3-硫酮的氧化
有文獻報道,2-[2-(1-氯環丙基)-3-(2-氯苯基)-2-羥基丙基]-1,2,4-三唑烷-3-硫酮可以通過在氫氧化鉀(KOH)及硫(S)的存在下加熱通氧來實現向丙硫菌唑的轉化[10]。但有機體系在加熱條件下通氧具有一定的危險性,對操作要求較高,同時該步收率也僅有70%左右。以三氯化鐵和雙氧水為氧化體系,反應在室溫下即可進行,操作簡便,收率在90%以上。
3 結論
在綜合分析和借鑒國內、外丙硫菌唑合成方法的基礎上,設計了丙硫菌唑合成工藝路線,該路線共4步,總收率達到53%。該工藝路線原料價廉易得,反應條件相對溫和,反應溶劑容易回收循環利用,中間體易于分離和純化等,具有較好的工業應用前景。
[1]關云飛,孫克,張敏恒.丙硫菌唑合成方法述評[J].農藥,2014,53 (9):696-698.
[2]Parker J E,Warrilow A G,Cools H J,et al.Mechanism of Binding of Prothioconazole to Mycosphaerella Graminicola CYP51 Differs from That of Other Azole Antifungals[J].Applied and Environmental Microbiology,2011,77(4):1460-1465.
[3]Thomas H,Udo K,Wolfgang K,et a1.Preparation of Benzyl Ketones and an Oxirane:US,5146001[P].1992-09-08.
[4]Metzger A,Schade M,Knochel P.LiCl-Mediated Preparation of Highly Functionalized Benzylic Zinc Chlorides[J].Organic Letters, 2008,10(6):1107-1110.
[5]王美娟,廖道華,曾仲武,等.丙硫菌唑的合成 [J].農藥,2009,48 (3):172-173;201.
[6]Hupperts A,Ruther M,Jautelat M.Method for Production of a Triazolinethione Derivative:WO,0146158[P].2001-06-28.
[7]Jautelat M,Erdman D.Process for Preparing Triazolinethione Derivatives:US,6l72236[P].2001-01-09.
[8]王哲清.簡述格氏反應 [J].中國醫藥工業雜志,2012,43(4): 311-316.
[9]Li J,Liao X,Liu H,et al.A New Way to Prepare Grignard Reagent from RX(X=Cl,Br)Using the Mixture of BrCH2CH2Br and I2as an Initiator[J].Synthetic Communications,1999,29(6):1037-1039.
[10]Jautelat M,Erdman D.Method for Producing Triazolinethion Derivatives:WO,9918087[P].1999-04-15.
(責任編輯:顧林玲)
Study on the Synthesis of Prothioconazole
MA Yi-chao,ZHANG Meng-meng,SHEN Guo-fu,TANG Bao-he,WANG Kuan,CHENG Yi-nan*,LI Hong-lian
(College of Plant Protection,Henan Agricultural University,Zhengzhou 450002,China)
Prothioconazole was prepared successfully with 2-chlorobenzyl chloride,2-chloro-1-(1-chlorocyclopropyl) ethanone and hydrazine hydrate,via Grignard reaction,hydrazine substitution,cyclization and oxidation.The total yield of the four synthetic steps reached 53%,and the product purity reached 95%.The procedure showed several advantages such as abundant raw materials,mild reaction conditions,easily recycled reaction solvents,easily purified intermediates and high product quality.It was suitable for industrial development.
prothioconazole;synthesis;Grignard reaction;substituted hydrazine

TQ 455.4+7
A
10.3969/j.issn.1671-5284.2017.04.004
2017-04-21;
2017-05-08
河南省科技攻關計劃項目(172102110042);公益性行業(農業)科研專項(201503112);河南省高等學校重點項目(16A210006)
馬藝超(1992—),男,河南省洛陽市人,碩士研究生,從事農藥開發與合成研究
程繹南(1970—),男,鄭州市人,副教授,博士,從事農藥、醫藥工程研究與教學。E-mail:chyn212@aliyun.com
