急性胰腺炎患者血清微管相關蛋白1輕鏈3水平變化及意義
2017-09-03 10:27:41吳海龍孫建兵王建祥
山東醫藥 2017年30期
吳海龍,孫建兵,王建祥
(武漢市普愛醫院,武漢430030)
急性胰腺炎患者血清微管相關蛋白1輕鏈3水平變化及意義
吳海龍,孫建兵,王建祥
(武漢市普愛醫院,武漢430030)
目的 觀察急性胰腺炎(AP)患者血清微管相關蛋白1輕鏈3(MAP1-LC3)水平變化,探討其臨床意義。方法 選擇輕型AP患者(MAP組)31例和重癥AP患者(SAP組)31例,于發病24 h內抽取血清樣本,另選擇31例健康者作為對照組。檢測血清淀粉酶(Ams)、MAP1-LC3水平,對AP患者的腹部CT進行評分。結果 與對照組比較,MAP組、SAP組血清Ams、MAP1-LC3水平及腹部CT評分均升高(P均<0.05),其中SAP組較MAP組上述指標升高(P均<0.05);血清MAP1-LC3水平與腹部CT評分呈正相關(r=0.977,P<0.01),與Ams呈正相關。結論 AP患者發病早期血清MAP1-LC3水平明顯升高,并與AP的病情嚴重程度呈正相關關系;血清MAP1-LC3可能是早期預測AP嚴重程度的有效標記物。
急性胰腺炎;自噬;微管相關蛋白1輕鏈3
急性胰腺炎(AP)起病急、變化快、并發癥多,病死率高達20%[1~4]。目前臨床上對AP的處理主要是對癥支持治療,仍缺乏特異、有效的治療措施。目前對AP發病的中心環節胰酶異常的激活機制尚不明確。研究認為,AP的病理基礎為胰酶原在胰腺細胞內異常的激活,從而引起胰腺的自我消化,進一步引起局部及全身炎癥反應[5~7]。自噬與AP的發生、發展密切相關,微管相關蛋白1輕鏈3(MAP1-LC3)是自噬體形成及胞質到空泡靶向囊泡必需的蛋白。本研究通過檢測AP患者的血清MAP1-LC3水平,分析血清MAP1-LC3水平與AP嚴重程度的關系,探討AP發病的機制。
1 資料與方法
1.1 臨床資料 選擇2016年1~12月我院收治的AP患者62例,男39例、女23例,年齡(68.2 ± 3.7)歲,均符合中華醫學會外科分會胰腺外科學組AP診治指南中的標準。分為輕型AP(MAP組)、重癥AP(SAP組)各31例。患者均在發病24 h內入院,既往無嚴重的心、腦、肺、腎、肝等基礎疾病,術前均未接受生物、免疫治療并有完整的臨床資料。另選擇性別、年齡相匹配的健康志愿者31例作為對照組。
1.2 血清淀粉酶(Ams)、MAP1-LC3水平檢測 采集AP患者發病24 h內及對照組空腹靜脈血標本,離心取上清液,分裝入EP管中,-80 ℃保存。使用全自動生化分析儀測定血清Ams水平;雙抗夾心酶聯免疫吸附(ELISA)法檢測血清MAP1-LC3水平,試劑盒購自武漢優爾生(USCN)科技股份有限公司。
1.3 腹部CT檢查方法 AP患者于入院24 h內行腹部增強CT檢查,采用改良的CT嚴重指數評分(MCTSI)標準評分。胰腺炎癥反應評分中,正常胰腺為0分,胰腺和(或)胰周炎性改變為2分,單發或多個積液區或胰周脂肪壞死為4分。胰腺壞死評分中,無胰腺壞死為0分,壞死范圍≤30%為2分,壞死范圍>30%為4分。以胰腺炎癥反應評分+胰腺壞死評分計算CT評分。
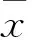
2 結果
2.1 三組血清Ams、MAP1-LC3水平比較 MAP組和SAP組血清Ams、MAP1-LC3水平均高于對照組,SAP組高于MAP組(P均<0.01)。
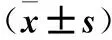
表1 三組血清Ams、MAP1-LC3水平比較
注:與對照組比較,*P<0.01;與MAP組比較,#P<0.01。
2.2 三組腹部CT評分比較 對照組、MAP組及SAP組腹部CT評分分別為(0.090±0.021)、(2.409±0.347)、(6.862±0.878)分,MAP組和SAP組腹部CT評分均高于對照組,SAP組高于MAP組(P均<0.01)。
2.3 血清MAP1-LC3與血清Ams、腹部CT評分的相關性 相關性分析顯示,血清MAP1-LC3與腹部CT評分呈正相關(r=0.977,P<0.01),與血清Ams呈正相關(r=0.765,P<0.05)。
3 討論
AP起病急、變化快、并發癥多、病死率高,一直是臨床治療的難點,及時、準確、高效地對AP患者進行合理診治尤為重要。多數研究認為,AP的病理基礎是胰酶原在胰腺細胞內非正常的激活,引起胰腺自我消化,進一步引起局部及全身炎癥反應。但胰腺細胞內的酶原具體是如何激活的,目前尚不完全清楚。AP時自噬的改變是近年的研究熱點。AP早期階段的特征性改變是胰腺細胞胞質內出現許多空泡[8],空泡中含有活化的胰酶。隨著自噬相關研究的進展,證實AP早期出現的空泡結構就是自噬空泡[9,10]。自噬與AP的發生、發展密切相關,過度自噬不僅導致AP早期的胰酶異常激活,還可介導炎癥的病理生理過程。過度自噬導致線粒破壞,引起線粒體活性氧簇產生增多,導致炎癥小體激活。
自噬相關信號轉導通路錯綜復雜。MAP1-LC3是自噬形成調控的主要因子之一,對自噬的發生和發展有著重要的調節作用。MAP1-LC3在自噬體的形成及膜來源中發揮作用[11,12]。有研究[13,14]通過對分離胰腺細胞、動物模型和人體組織的研究,發現AP時自噬量受損,胰腺腺泡內大量異常自噬空泡堆積;自噬的異常增多是導致酶原的激活的誘因,認為自噬導致胰腺腺泡細胞空泡的形成以及酶原的激活,最終誘發了AP的形成。MAP1-LC3是自噬調控的重要分子機制之一,MAP1-LC3的表達與自噬相互促進,血清MAP1-LC3水平增高可以間接反映AP患者自噬的過度激活。AP的發病與過度自噬密切相關,而MAP1-LC3是自噬的重要調控蛋白。本研究發現,SAP組血清Ams水平高于MAP組,MAP組、SAP組發病早期血清MAP1-LC3水平明顯升高,且SAP組較MAP組血清MAP1-LC3水平增高;AP患者的血清MAP1-LC3水平和血清Ams腹部CT評分呈正相關,提示AP患者發病早期血清MAP1-LC3水平與AP的病情嚴重程度呈正相關,血清MAP1-LC3可能是早期預測AP嚴重程度的有效標記物。因此可以通過減少MAP1-LC3表達而抑制胰腺腺泡細胞自噬的發生,從而減少胰蛋白酶原的激活以及AP時胰腺組織水腫、出血及壞死的發生[15],MAP1-LC3可能作為藥物治療AP的特異性靶點之一。
綜上所述,AP患者發病早期血清MAP1-LC3水平明顯升高,并與AP的病情嚴重程度呈正相關,MAP1-LC3可能是早期預測AP嚴重程度的有效血清標記物。
[1] Tomas RG, Alberto GC, Javier GV. Etiology of acute pancreatitis[J]. Cent Eur J Med, 2014,9(4):530-542.
[2] Singh VK, Moran RA, Afghani E, et al. Treating acute pancreatitis: what′s new[J]. Expert Rev Gastroenterol Hepatol, 2015,9(7):901-911.
[3] Wu BU, Bakker OJ, Papachristou GI, et al. Blood urea nitrogen in the early assessment of acute pancreatitis: an international validation study[J]. Arch Intern Med, 2011,171(7):669-676.
[4] Mallédant Y, Malbrain ML, Reuter DA. What′s new in the management of severe acute pancreatitis[J]. Int Care Med, 2015,41(11):1957-1960.
[5] 中華醫學會外科學分會胰腺外科學組.急性胰腺炎診治指南(2014版)[J].全科醫學臨床與教育,2015,14(3):1-5.
[6] Mareninova OA, Hermann K, French SW, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis[J]. J Clin Invest, 2009,119(11):3340-3355.
[7] Gukovskaya AS, Gukovsky I. Autophagy and pancreatitis[J]. Am J Physiol Gastrointest Liver Physiol, 2012,303(9):993-1003.
[8] Van Acker GJ, Weiss E, Steer ML, et al. Cause-effect relationships between zymogen activation and other early events in secretagogue-induced acute pancreatitis[J]. Am J Physiol Gastrointest Liver Physiol, 2007,292(6):1738-1746.
[9] Lerch MM, Gorelick FS. Early trypsinogen activation in acute pancreatitis[J]. Med Clin North Am, 2000,84(3):549-563.
[10] Watanabe O, Baccino FM, Steer ML, et al. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis[J]. Am J Physiol, 1984,246(4):457-467.
[11] Jacob TG, Sreekumar VI, Roy TS, et al. Electron-microscopic evidence of mitochondriae containing macroautophagy in experimental acutepancreatitis: implications for cell death[J]. Pancreatology, 2014,14(6):454-458.
[12] Xu B, Bai B, Sha S, et al. Interleukin-1β induces autophagy by affecting calcium homeostasis and trypsinogen activation in pancreatic acinar cells[J]. Int J Clin Exp Pathol, 2014,7(7):3620-3031.
[13] Zhu H, Huang L, Zhu S, et al. Regulation of autophagy by systemic admission of microRNA-141 to target HMGB1 in l-arginine-induced acute pancreatitis in vivo[J]. Pancreatology, 2016,16(3):337-346.
[14] Ohmuraya M, Yamamura K. Autophagy and acute pancreatitis: a novel autophagy theory for trypsinogen activation[J]. Autophagy, 2008,4(8):1060-1062.
[15] Hashimoto D, Ohmuraya M, Hirota M, et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells[J]. J Cell Biol, 2008,181(7):1065-1072.
10.3969/j.issn.1002-266X.2017.30.028
R657.5
B
1002-266X(2017)30-0088-02
2017-03-15)
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
中老年保健(2021年3期)2021-08-22 06:50:04
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年12期)2019-05-21 02:55:32
學苑創造·A版(2015年11期)2016-01-14 09:03:27
現代檢驗醫學雜志(2014年4期)2014-02-02 02:44:59
