不同價態五氧化二釩吸附水和氨的機理研究
2017-09-18 08:24:58高正陽梁冉冉范軍輝
動力工程學報 2017年9期
關鍵詞:催化劑
高正陽, 梁冉冉, 范軍輝
(華北電力大學 能源動力與機械工程學院,河北保定 071003)
不同價態五氧化二釩吸附水和氨的機理研究
高正陽, 梁冉冉, 范軍輝
(華北電力大學 能源動力與機械工程學院,河北保定 071003)
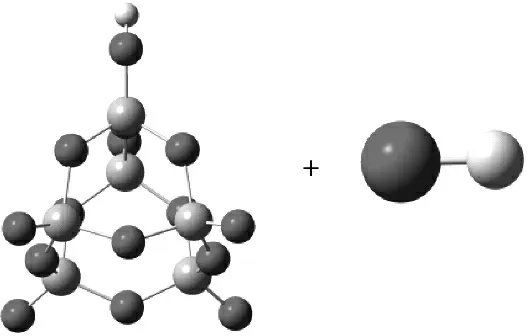
為研究不同價態的V2O5(V6O15及V6O15+)吸附H2O和NH3的反應機理,應用量子化學得到反應物、中間體、過渡態和產物的幾何構型,對比分析反應過程中優化結構的勢能、反應能壘、鍵長和吸附能等數據.結果表明:V6O15和V6O15+均可吸附H2O生成V—OH,但是對于后者整個反應表現為放熱,而且反應能壘更低,說明V6O15+更容易與H2O反應生成Br?nsted酸位;NH3在V6O15+上的吸附能更大,更容易形成可與NO反應的—NH4+;在SCR脫硝反應中陽離子團簇V6O15+比中性團簇V6O15活性更大.
五氧化二釩; 吸附; 量子化學; 反應機理
氮氧化物(NOx)是造成酸雨和城市大氣污染的主要原因.近年來,中國燃煤電廠產生的NOx約占排放總量的60%,隨著燃煤電廠數量的不斷增加,NOx排放量及其所占比例還將繼續增加[1].為響應世界各國保護環境及人類生命財產安全的號召,我國制定了嚴格的排放標準來控制火電廠NOx的排放量[2].目前,選擇性催化還原(SCR)被認為是一種最有效的煙氣脫硝方法[3].SCR的工作原理是向鍋爐排放的攜帶有大量NOx的煙氣中噴入NH3等還原劑,在脫硝催化劑的作用下NH3等還原劑與煙氣中的NOx反應,生成對大氣無污染的N2和H2O.
煙氣SCR脫硝工藝的技術核心是催化劑.脫硝催化劑一般由活性組分和載體組成,以V2O5為主要活性組分、TiO2為載體的催化劑是當前工業脫硝催化劑的主流劑種.國內外學者對釩系催化劑脫硝反應機理的描述不盡相同,歸納起來認為NH3-SCR脫硝反應主要有2種機理[4]:Eley-Rideal機理(E-R機理)和Langmuir-Hinshelwood機理(L-H機理).Topsoe等[5-6]提出SCR脫硝反應的E-R和L-H結合機理,被廣泛認可,該機理涉及2種V活性位,即Br?nsted位(吸附位V—OH)和Lewis位(氧化位V=O).NH3吸附在Br?nsted酸位(B酸位)上形成的—NH4+在SCR脫硝反應機理中起關鍵作用,氣相中的NO與活化的—NH4+發生反應并生成中間體,分解成N2和H2O[7].V2O5結構本身只有Lewis位,—NH4+在V—OH基上形成,B酸位由V=O吸附水轉化形成[8].因此,B酸位的形成以及NH3在B酸位的吸附對于研究SCR脫硝反應至關重要.
為了在分子層次上揭示催化反應的相關機理,眾多學者對過渡金屬氧化物團簇和H2O、NH3分子反應進行了研究[9-10].相比于V2O5中性團簇,針對V2O5陽離子團簇對B酸位形成的影響以及NH3吸附的研究還很少.Ma等[11]研究了H2O分子吸附在V4O10陽離子團簇并生成V—OH的過程,但是沒有探討V4O10中性團簇吸附H2O分子并生成V—OH的過程.關于V2O5中性團簇和陽離子團簇吸附水形成羥基及吸附氨過程的研究也很少.筆者利用密度泛函理論(DFT)結合精度更高的V6O15、V6O15+簇模型,模擬Lewis位吸附H2O分子形成吸附位V—OH的微觀機理,得到了反應過程中形成的中間體和過渡態的結構,進一步探討NH3在B酸位的吸附,對比分析不同價態的團簇模型在脫硝過程中的優劣性,對提升催化劑的脫硝效率有一定參考意義.
1 計算方法
1.1理論方法和反應機理
隨著計算機的發展,量子化學逐漸成為探討微觀反應機理的重要手段[12].密度泛函理論具有良好的計算速度和精確度,因此被廣泛地應用于量子化學計算[13].筆者采用密度泛函理論B3LYP,體系中的金屬原素V選擇lanl2dz基組,除V以外的H、O和N等元素處于多電荷體系,采用單純的標準基組不能滿足計算精度,因此選擇6-311g(d,p)基組.在指定的基組水平上采用雙雜化泛函方法得到反應中所有物質的能量.計算采用Gaussian09軟件包.
由于吸附水形成羥基過程的反應物不同,研究反應過程也會有差異.
反應Ⅰ,V6O15吸附水分子:
V6O14(OH)2
(1)
反應Ⅱ,V6O15+吸附水分子:
(2)
應用量子化學計算V6O15和V6O15+團簇吸附水反應過程中涉及到的反應物、中間體、過渡態和產物的幾何構型,并對其進行振動頻率分析,確保優化后都是能量最低的結構.在相關幾何參數的基礎上進行熱能和零點能校正,從而得到反應活化能.構造B酸位后模擬計算NH3在V6O15和V6O15+團簇羥基上的吸附,對比分析優化后原子間鍵長的變化和吸附能的大小,獲得不同價態釩基催化劑吸附氨形成—NH4+的能力.
1.2吸附能的計算方法
通過吸附能來衡量催化劑表面對吸附質的作用,吸附能(Eads)為吸附前后體系的能量變化:
(3)
式中:E(A)和E(B)分別為吸附前原子簇模型和吸附質的能量;E(AB)為吸附后體系的能量.
吸附能為負值表示可能發生吸附,否則吸附不會發生.Eads負值越大,吸附能力越強,反應越容易發生.
2 結果與分析
2.1V2O5吸附H2O形成B酸位
2.1.1 H2O吸附生成—OH的過程
采用密度泛函理論B3LYP,調整指定鍵長后掃描勢能面,將勢能面上的鞍點作為初始結構,在6-311g(d,p)/lanl2dz基組水平上進行結構優化,得到整個反應過程,如圖1和圖2所示.

+→→→
R IM1 TS1 P1
圖1 V6O15吸附H2O的過程
Fig.1 Reaction process of H2O adsorption by V6O15

R IM2 TS2 IM3
P
圖2 V6O15+吸附H2O的過程

圖1給出了中性團簇V6O15吸附H2O生成B酸位的過程,H2O分子中的一個H原子和V=O(1)的O(1)原子結合,該反應存在中間體和過渡態.在整個反應過程中,H—O(1)兩原子間的距離逐漸縮小(∞→0.283 nm→0.116 nm→0.098 nm,∞表示原子間的距離不足以成鍵),反映了H—O(1)鍵的形成過程.釩基上V=O(1)雙鍵距離逐漸拉長(0.157 nm→0.157 nm→0.169 nm→0.174 nm),V原子與O(1)原子間的作用力變弱,有利于脫離H2O分子后的H原子靠近O(1)形成B酸位.過渡態頻率分別為-1 553.90 cm-1、33.21 cm-1和35.97 cm-1等,有且僅有一個虛頻.在虛頻振動形式下,H原子向O(1)原子方向有明顯的振動,有利于發生反應,說明存在該過渡態,筆者研究的機理是可信的.
陽離子團簇V6O15+吸附H2O生成B酸位的過程如圖2所示.H2O分子吸附在V6O15+上生成穩定的中間體IM2,經過渡態TS2得到中間體IM3,優化后H2O分子分解,其中一個H原子逐漸遠離并靠近O(1)原子,生成V6O15H+和OH.在反應過程中,H—O(1)鍵的距離逐漸縮小(∞→0.243 nm→0.129 nm→0.103 nm→0.098 nm),反映了H—O(1)鍵的形成過程.在整個過程中,所有經過優化的中間體振動頻率均為正,過渡態有且僅有一個虛頻,頻率為-1 159.98 cm-1.在虛頻振動形式下,H原子沿反應途徑遠離H2O分子并向釩基O(1)原子方向有明顯的振動,說明該過渡態的存在是合理的.
2.1.2 沿反應途徑(IRC)體系的勢能變化
分別以反應狀態的反應途徑和總能量為x軸和y軸作圖,得到沿反應途徑(IRC)體系的勢能變化曲線,如圖3和圖4所示.

圖3 V6O15吸附H2O沿反應途徑(IRC)的能量變化
Fig.3 Energy changes of adsorption of H2O on V6O15neutral clusters along intrinsic reaction coordinate (IRC)

圖吸附H2O沿反應途徑(IRC)的能量變化
由圖3可知,反應物的能量低于產物能量,說明是吸熱反應.在H2O分子靠近釩基并形成V—OH的過程中,當H2O分子中O—H鍵逐漸斷裂,H原子遠離羥基并將要與V6O15中的V=O(1)成鍵時,形成過渡態TS1,反應體系吸收熱量;當H原子與O(1)成鍵形成V—OH時,反應體系放出熱量.
由圖4可知,反應物的能量高于產物能量,說明是放熱反應.當H2O分子中的一個H原子逐漸遠離并將要與V6O15+中的V=O(1)成鍵時,形成過渡態TS2,過程吸收熱量;當H原子與O(1)成鍵形成V—OH時,反應體系放出熱量.
2.1.3 反應過程中的能量
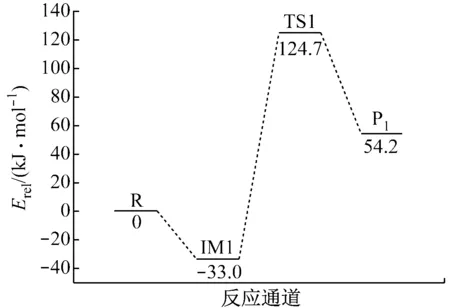
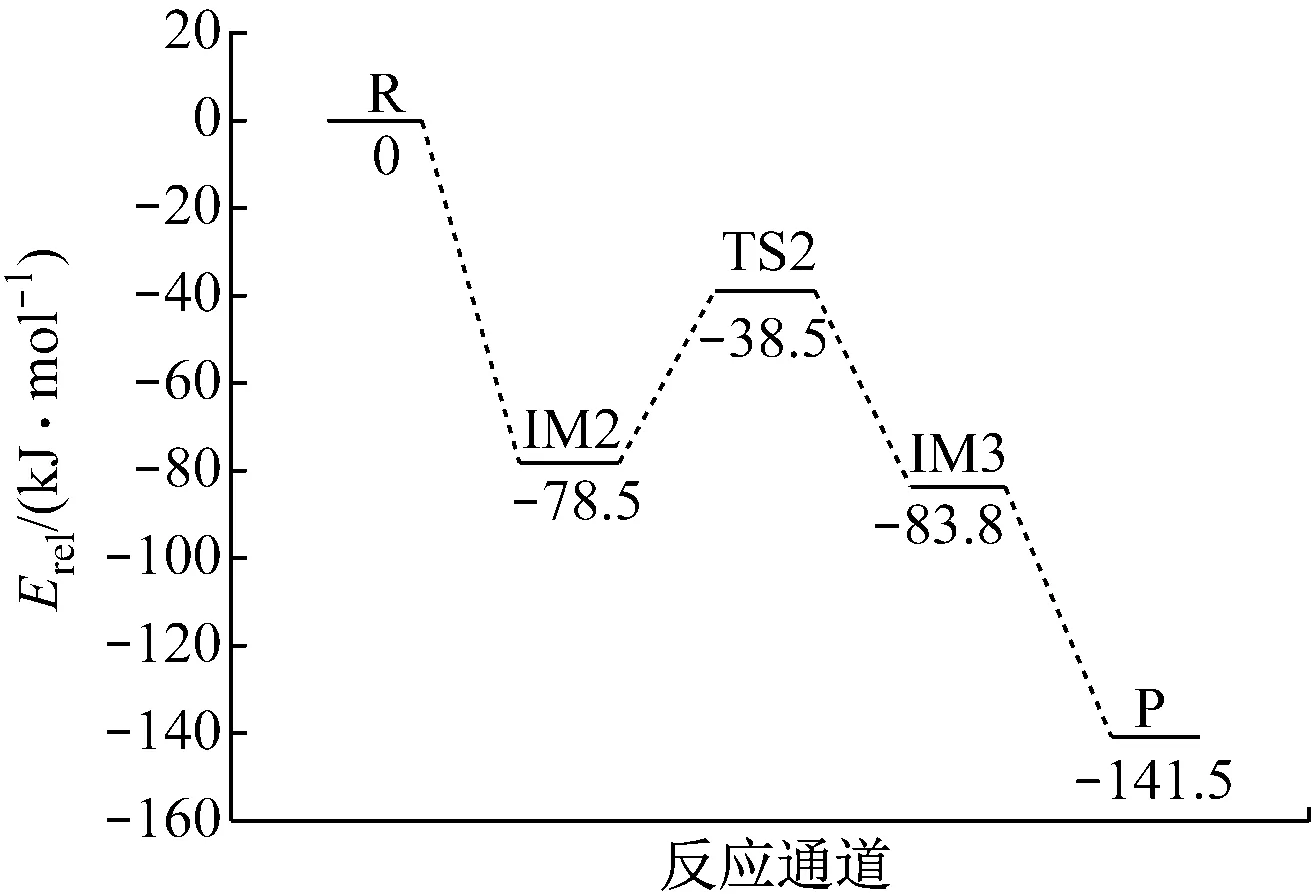
V2O5吸附H2O生成B酸位的反應涉及到各駐點(反應物R、過渡態TS、中間體IM以及產物P)的總能量、相對能量和過渡態結構的虛頻,如表1和表2所示.根據表1和表2中數據繪制各駐點相對能量的變化,如圖5所示.
由表1、表2和圖5可知,中性團簇V6O15吸附H2O生成B酸位的反應能壘為157.7 kJ/mol.陽離子團簇V6O15+吸附H2O生成羥基的反應能壘為40.0 kJ/mol.
表1V6O15吸附H2O的過程中各駐點結構的單點能E0、零點校正能EZPE、總能量Etot、相對能Erel和虛頻ν
Tab.1E0,EZPE,Etot,Erelofvariouscompoundsandtheimaginaryfrequencyoftransitionstates(ν)inreactionprocessofH2OadsorptionbyV6O15

各駐點結構E0/(MJ·mol-1)EZPE/(MJ·mol-1)Etot/(MJ·mol-1)Erel/(kJ·mol-1)ν/cm-1V6O15-4085.52480.1567-4085.3681H2O-200.60660.0557-200.5512R(V6O15+H2O)-4285.91920IM1-4286.17520.2226-4285.9523-33.0207TS1-4286.00460.2098-4285.7948124.6729-1553.90P1(V6O14(OH)2)-4286.08250.2177-4285.865154.1887
表2V6O15+吸附H2O的過程中各駐點結構的單點能E0、零點校正能EZPE、總能量Etot、相對能Erel和虛頻ν

各駐點結構E0/(MJ·mol-1)EZPE/(MJ·mol-1)Etot/(MJ·mol-1)Erel/(kJ·mol-1)ν/cm-1V6O15+-4084.46360.1518-4084.3118H2O-200.60660.0554-200.5512R(V6O15++H2O)-4284.86270IM2-4285.16020.2190-4284.9415-78.5489TS2-4285.10140.2001-4284.9013-38.4554-1159.98IM3-4285.15520.2085-4284.9467-83.8443V6O15H+-4086.30930.1804-4086.1289OH-198.89580.0205-198.8753P(V6O15H++OH)-4285.0042-141.4730
根據對IRC勢能變化和反應能壘大小的分析,H2O分子在陽離子團簇吸附形成B酸位時為放熱反應,反應能壘較低.因此,相比于中性團簇V6O15,V6O15+更容易吸附H2O生成—OH,反應活性更大.
2.2NH3在V2O5羥基上的吸附
(a) V6O15吸附H2O的反應過程
(b) V6O15+吸附H2O的反應過程
NH3在V2O5表面的吸附位主要有Lewis位和B酸位,在SCR脫硝反應過程中E-R機理和L-H機理均可能發生,NO以弱吸附物種形式參與反應的可能性更大.NH3吸附在催化劑酸性位V—OH形成—NH4+,—NH4+與氣相中的NO結合生成中間體,分解成N2和H2O.因此,探討NH3吸附在催化劑表面V—OH形成—NH4+的過程對于研究SCR脫硝反應機理具有重要意義.筆者研究了NH3在不同價態V2O5(V6O15和V6O15+)表面B酸位的吸附情況,如圖6所示.表3給出了NH3在V6O15和V6O15+表面B酸位吸附前后原子間鍵長的變化及吸附能的大小.其中d為鍵長,Eads為吸附能.

(a) NH3吸附在V6O15表面B酸位的模型

(b) NH3吸附在V6O15+表面B酸位的模型
表3NH3在V2O5團簇B酸位吸附后的鍵長和吸附能(括號內為吸附前數值)
Tab.3AdsorptionparametersforNH3adsorbedatBr?nstedsiteforV2O5cluster(valuesinbracketarethatbeforeadsorption)

吸附構型dH—N/nmdO—H/nmEads/(kJ·mol-1)V6O15—NH30.1470.109(0.097)-89.396V6O15+—NH30.1070.163(0.098)-158.408
由表3可知,NH3在V6O15和V6O15+表面B酸位的吸附能Eads分別為-89.396 kJ/mol和-158.408 kJ/mol,吸附能絕對值均大于發生化學吸附時的能量(30 kJ/mol),即發生了化學吸附,陽離子團簇對氨的吸附能遠大于中性模型.從吸附能大小可以看出,SCR脫硝反應中V6O15+比V6O15更容易吸附NH3.V6O15吸附NH3時O(1)—H鍵長為0.109 nm,O(1)—H與氮原子的距離為0.147 nm.陽離子構型上O(1)—H鍵被拉長0.066 nm,H與N原子的距離為0.107 nm,與NH3中N—H鍵長計算值0.102 nm十分接近,說明V6O15+表面B酸位上NH3與羥基更容易作用生成—NH4+.
綜上表明,NH3既能吸附在V6O15表面B酸位上生成—NH4+,也能在V6O15+的B酸位上生成—NH4+,后者吸附能更大、更穩定,O(1)H與N原子的距離與NH3中N—H鍵長更接近.對于多聚合態的V2O5,在SCR脫硝反應中NH3與羥基作用生成的—NH4+有重要作用,因此陽離子團簇V6O15+更容易吸附氨生成—NH4+,活性更大.
3 結 論
(1)V6O15和V6O15+均可吸附H2O生成V—OH,后者整個反應表現為放熱,反應能壘更低,說明V6O15+更易與H2O反應生成B酸位.
(2)NH3在V6O15和V6O15+表面B酸位上的吸附均為化學吸附,但是在V6O15+上的吸附能更大,O(1)H與N原子的距離更小,更容易形成可與NO反應的—NH4+.
(3)在SCR脫硝反應中陽離子團簇V6O15+比中性團簇V6O15的活性更大.
[1] 朱禮想. 燃煤電廠氮氧化物(NOx)脫除技術方法探討和應用[J].價值工程, 2015(12): 64-66.
ZHU Lixiang. Discussion and application of NOxremoval technology in coal-fired power plant[J].ValueEngineering, 2015(12): 64-66.
[2] 高婕, 王禹, 張蓓. 我國大氣氮氧化物污染控制對策[J].環境保護科學, 2004, 30(5): 1-3.
GAO Jie, WANG Yu, ZHANG Bei. Countermeasure of atmospheric nitrogen oxide pollution in China[J].EnvironmentalProtectionScience, 2004, 30(5): 1-3.
[3] 譚青, 馮雅晨. 我國煙氣脫硝行業現狀與前景及SCR脫硝催化劑的研究進展[J].化工進展, 2011, 30(增刊1): 709-713.
TAN Qing, FENG Yachen. Present status and perspective of China's flue gas denitration industry and research progress of SCR catalysts[J].ChemicalIndustryandEngineeringProgress, 2011, 30(S1): 709-713.
[4] 高慧聰. V4+/V5+比值調變與SCR脫硝活性及催化劑性能[D]. 哈爾濱: 哈爾濱工程大學, 2013.
[5] TOPSOE N Y, DUMESIC J A, TOPSOE H. Vanadia-titania catalysts for selective catalytic reduction of nitric-oxide by ammonia: I.I. Studies of active sites and formulation of catalytic cycles[J].JournalofCatalysis, 1995, 151(1): 241-252.
[6] TOPSOE N Y, TOPSOE H, DUMESIC J A. Vanadia/titania catalysts for selective catalytic reduction (SCR) of nitric-oxide by ammonia: I. combined temperature-programmed in-situ FTIR and on-line mass-spectroscopy studies[J].JournalofCatalysis, 1995, 151(1): 226-240.
[7] 杜學森. 鈦基SCR脫硝催化劑中毒失活及抗中毒機理的實驗和分子模擬研究[D]. 杭州: 浙江大學, 2014.
[8] 姜燁. 鈦基SCR催化劑及其鉀、鉛中毒機理研究[D]. 杭州: 浙江大學, 2010.
[9] 彭吉偉. 選擇性催化還原脫硝催化劑反應機理與失活分析[D]. 濟南: 山東大學, 2014.
[10] 孫克勤. 選擇性催化還原脫硝的理論及實驗研究[D]. 南京: 南京理工大學, 2007.
[11] MA Jiabi, ZHAO Yanxia, HE Shenggui, et al. Experimental and theoretical study of the reactions between vanadium oxide cluster cations and water[J].TheJournalofPhysicalChemistryA, 2012, 116(9): 2049-2054.
[12] TYRRELL J, KAR T, BARTOLOTTI L J. A study of the mechanism of the reaction between ozone and the chlorine atom using density functional theory[J].TheJournalofPhysicalChemistryA, 2001, 105(16): 4065-4070.
[13] 高正陽, 呂少昆, 李晉達, 等. 褐煤表面吸附水分子的微觀機理[J].動力工程學報, 2016, 36(4): 258-264, 293.
GAO Zhengyang, Lü Shaokun, LI Jinda, et al. Micro-mechanism of water molecule adsorption on lignite surfaces[J].JournalofChineseSocietyofPowerEngineering, 2016, 36(4): 258-264, 293.
Mechanism Study of H2O and NH3Adsorption by V2O5with Different Valences
GAOZhengyang,LIANGRanran,FANJunhui
(School of Energy, Power and Mechanical Engineering, North China Electric Power University,Baoding 071003, Hebei Province, China)

V2O5; adsorption; quantum chemistry calculation; reaction mechanism
2016-09-18
:2017-01-04
中央高校基本科研業務費專項資金資助項目(10MG19)
高正陽(1972-),男,河北保定人,副教授,博士,主要從事煤中有害痕量元素遷移規律與控制方法方面的研究. 電話(Tel.):18331125675;E-mail:liangran0128@163.com.
1674-7607(2017)09-0738-06
:TK122
:A
:470.10
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
