光誘導約束刻蝕體系中的TiO2納米管陣列光電極上Cu的沉積及抑制
2017-11-01 18:11:05黃雅鈺方秋艷周劍章詹東平田中群
物理化學學報 2017年10期
黃雅鈺 方秋艷 周劍章 詹東平 時 康 田中群
(廈門大學化學化工學院化學系, 固體表面化學國家重點實驗室, 福建 廈門 361005)
光誘導約束刻蝕體系中的TiO2納米管陣列光電極上Cu的沉積及抑制
黃雅鈺 方秋艷 周劍章*詹東平 時 康 田中群
(廈門大學化學化工學院化學系, 固體表面化學國家重點實驗室, 福建 廈門 361005)
光誘導約束刻蝕可作為一種無應力的化學平坦化方法用于Cu的拋光。我們發現在光誘導約束刻蝕工件Cu的過程中,工具表面的TiO2納米管上可能出現Cu沉積。通過掃描電子顯微鏡及其能譜,X射線光電子能譜等方法分析其沉積形貌和成分組成,探究在工具–工件之間的微納尺度液層中Cu光催化還原沉積的機制,并在模擬液中研究Cu沉積對刻蝕體系的影響。探究引入攪拌、加入絡合劑對TiO2納米管表面Cu的沉積的抑制,并考察抑制措施對于工件Cu刻蝕的影響。結果表明Cu沉積會增強TiO2納米管光電極的光催化性能,但隨著沉積量的增加,增強機制會發生變化;在嘗試抑制Cu沉積時也發現改善傳質以抑制Cu沉積的同時也會帶來工件Cu的刻蝕增強;采用添加絡合劑結合改善傳質的方法有望在抑制Cu沉積的同時提高平坦化效果。所以抑制方法和條件的選擇需兼顧對工具–工件之間微納液層中的多個化學和傳質過程的影響。這些研究對于進一步優化光誘導約束刻蝕體系及其在化學平坦化中的應用有重要的指導意義。
光誘導約束刻蝕;TiO2納米管陣列;羥基自由基;約束劑;Cu的光催化沉積;傳質
1 引 言
田昭武院士提出的約束刻蝕劑層技術具有有效控制刻蝕程度和對距離敏感的特點,已在半導體、金屬及合金等材料上實現了三維超微圖形的復制加工1?3。我們課題組在此基礎上提出了一種新的無應力拋光技術—光誘導約束化學平坦化方法,并應用于銅(Cu)的平坦化4。其基本原理是:加工工具表面的TiO2納米管光催化劑層在光誘導下產生刻蝕劑羥基自由基(?OH),溶液中的約束劑(H2O、甘氨酸等)與?OH發生清除反應,使?OH的擴散受限而被約束在表面的微納米尺度中,形成約束刻蝕劑層;當逼近工具的工件Cu的突起部分進入約束刻蝕劑層時將發生化學刻蝕溶解,而未進入約束刻蝕劑層的部分則基本不發生反應,從而實現Cu表面平坦化的效果。
但是在光誘導約束刻蝕Cu的過程中,我們觀察到在工具表面的TiO2納米管光催化劑層上可能出現Cu的沉積。文獻中已有一些在TiO2表面光催化沉積Cu的報道。如Montini等5在TiO2表面光催化沉積Cu以制備活潑和廉價的納米復合物。Lv等6在TiO2光催化劑層表面光沉積Cu納米粒子以獲得可以和TiO2表面修飾Pt媲美的光催化劑。Canterino等7研究了Cu(II)在有機物種存在的情況下,作為犧牲劑而在TiO2表面光催化沉積的機制。還有一些關于TiO2表面修飾、摻雜Cu及其氧化物以改變其光催化性能的報道。如Wu等8報道了在銳鈦礦晶型的TiO2表面沉積Cu的粒子(介于0和+1價之間)會大大提高TiO2從甲醇水溶液中制氫的光催化活性。Tseng等9報道適量地沉積Cu2O及單質Cu可以增加活性位點的數量從而提高光催化的效率,但當沉積位點更多時,單位時間內單位催化活性位點上的轉化數將下降,即意味著光催化活性的下降。并且過量的沉積也會帶來TiO2表面的遮擋,減小光催化的能力。那么在光誘導約束刻蝕體系中,TiO2納米管光催化劑層表面的Cu是如何沉積的?沉積的Cu或其氧化物是否會對TiO2納米管的光催化活性有影響,是否會影響刻蝕劑?OH的生成,進而對光誘導約束刻蝕和平坦化產生影響?如何抑制TiO2納米管表面Cu的沉積?因此,很有必要深入探究光誘導約束刻蝕體系中工具表面的TiO2納米管表面光催化沉積Cu的生成機制以及研究Cu沉積對光誘導約束刻蝕的影響。
本文著眼于研究光誘導約束刻蝕體系中 TiO2納米管光電極上光催化還原 Cu的沉積機制及其對刻蝕體系的影響,并嘗試添加絡合劑和增加對流抑制Cu的沉積,探究優化光誘導約束刻蝕的條件參數對約束刻蝕體系各個過程的影響。
2 實驗部分
2.1 試劑與儀器
試劑:六次甲基四胺(化學純,廣州化學試劑廠);Zn(NO3)2?6H2O、甘氨酸、硼酸、尿素、氨水、無水乙醇、丙酮、乙二胺四乙酸(EDTA)、硝酸銅(分析純,國藥集團化學試劑有限公司);六氟鈦酸銨(98%,Aldrich公司);FTO(武漢格奧儀器公司代理,美國 LOF公司生產);Cu片(99.9%,北京翠鉑林有色金屬技術開發中心)。所有的試劑沒有進一步純化。所用的溶液采用超純水(電阻率 18.2 M??cm)配制。
儀器:光照光源采用北京賽凡光電的 500 W氙燈和北京卓立漢光150 W氙燈;電鏡測試采用日本Hitachi公司的S4800 FESEM型掃描電鏡,能譜分析采用該儀器所配的X射線能譜分析(EDS)附件;X射線衍射測定采用荷蘭帕納科公司的Panalytical X?Pert PRO X射線衍射儀。Cu靶,掃描步長0.02°;X射線光電子能譜采用英國VG公司生產的ESCA LAB MK-Ⅱ型,Al Kα為X射線源,以C 1s的284.5 eV峰為參考;吸收光譜采用美國Varian公司生產的Cary 5000紫外-可見-近紅外分光光度計,測試時都扣除 FTO基底的吸收;暫態光電流響應采用上海辰華儀器公司生產的CHI-630a電化學工作站;熒光測試采用日本Hitachi公司的F-7000型熒光光譜儀。
2.2 實驗過程
TiO2納米管(NTs)陣列的制備:FTO導電玻璃依次在丙酮、乙醇和三次去離子水中超聲清洗兩次,氮氣吹干備用。采用恒電流陰極還原法制備ZnO納米棒,工作電極為FTO電極(10 mm × 20 mm),對電極為 Pt片電極(10 mm × 20 mm),兩電極平行相距10 mm,電解液為硝酸鋅(5 mmol?L?1Zn(NO3)2)和六次甲基四胺(5 mmol?L?1C6H12N4)的混合溶液,沉積電流密度為 0.25 mA?cm?2,沉積溫度控制在90 °C,沉積時間為20 min。用液相沉積的方法,將制備好的ZnO納米棒陣列浸入摩爾比1 : 3的六氟鈦酸銨和硼酸的混合溶液中4 h,液相沉積制備 TiO2納米管陣列。最后將制備好的TiO2納米管陣列在450 °C下煅燒2 h,升溫速率5 °C?min?1。
光誘導約束刻蝕Cu的實驗過程:將TiO2納米管陣列(10 mm × 10 mm)、100 μm 厚的聚四氟乙烯薄膜墊片(PTFE spacer)和 Cu 片(10 mm × 10 mm)用兩片帶螺孔的厚聚四氟乙烯固定座(厚度 2.2 mm)固定,其中一片聚四氟乙烯中間有一個8 mm ×8 mm的正方形孔允許光通過,具體結構示意如圖1所示。將固定好的整個裝置放入一個圓柱形電解池中,光從上向下照射,光照時間為4 h,光強為1.58 W?cm?2。本文中聚四氟乙烯薄膜墊片為十字型。刻蝕實驗中TiO2納米管陣列及Cu片上被墊片所遮擋的區域稱為遮擋區,未遮擋部分稱為鏤空區。只有在外置了攪拌的實驗中,磁子轉速為0–1000 r?min?1,其余均無外置攪拌。
模擬液中光催化還原沉積Cu的實驗過程:模擬液為1% (w)甘氨酸和1.25% (w) Cu(NO3)2,溶液pH ~ 2.5,加硝酸調整后為 pH ~ 5.8。將 TiO2納米管陣列置于該混合模擬溶液中,置于500 W氙燈下,在1.58 W?cm?2的光強下光照不同的時間。取出Cu/TiO2納米管陣列電極,用三次水沖洗,立即用氮氣吹干。
(1)給出輸入信號x(t),設置迭代次數,通常情況下,迭代次數越高,分解越精確,但是同時所花時間也越長。將重建信號初始化置零。

圖1 光誘導約束刻蝕Cu實驗裝置Fig.1 Experimental installation of photoinduced confined etching of Cu.
熒光測試實驗:選用熒光分析法對游離的·OH進行定量檢測。以無熒光特性的對苯二甲酸(TA)為分子探針,它與?OH反應后生成專一穩定且具有很強熒光特性的產物 2-羥基對苯二甲酸(TAOH)。通過測量424 nm處產物TAOH的熒光峰強度可以間接測出?OH的濃度。將TiO2納米管陣列放入自制石英光電解池中,溶液為1 mmol?L?1的對苯二甲酸(TA)和 0.01 mol?L?1的 NaOH,體積為3 mL。用150 W氙燈在35 mW?cm?2的光強下光照60 min,放置2 h后進行熒光測試。熒光測試的激發波長為312 nm。
暫態光電流響應測試:以TiO2納米管陣列或Cu/TiO2納米管陣列為工作電極,Pt絲為對電極,飽和甘汞電極(SCE)為參比電極,測試體系為0.01 mol?L?1NaOH 溶液,測試光強為 35 mW?cm?2。
3 結果與討論
3.1 光誘導約束刻蝕Cu后TiO2納米管的表征
光誘導約束刻蝕后,我們發現在工具表面的TiO2納米管上的鏤空區局部會出現肉眼可見的紫銅色。為確定該物質的形貌和組分,我們采用掃描電子顯微鏡(SEM)及掃描能譜(EDS)對該紫銅色區域進行表征。如圖 2的 SEM 所示,刻蝕后的TiO2納米管表面上覆蓋了一些由許多小顆粒堆積而成的大顆粒(主要是集中在納米管頂端),直徑可達微米級別。由圖2的EDS圖可見,刻蝕前TiO2納米管表面有Ti、O、Sn、C等信號,其中Sn信號主要來自基底 FTO,C等信號來自儀器和樣品表面的少量污染;而刻蝕后的 TiO2納米管表面,除了Ti和O信號外還多出了較強的Cu信號,由此可推測SEM圖中的大顆粒狀物質為Cu或者Cu的氧化物。
為進一步確認刻蝕后 TiO2納米管上沉積的Cu的價態,對其進行了XPS分析。圖3是刻蝕后TiO2納米管表面鏤空區的XPS譜。從圖3(a)的Cu 2p譜圖可知,Cu 2p3/2和Cu 2p1/2分別為932.4和952.3 eV,與單質Cu相符,與CuO(其Cu 2p3/2峰值為933.7 eV)不符,且沒有出現特征的CuO的震激伴峰,這說明了 TiO2納米管表面不存在 CuO。但Cu和Cu2O的2p3/2峰位置相近(Cu為932.6 eV,Cu2O為932.5 eV),因此我們進一步通過Cu LMM俄歇譜區分表面的氧化狀態。從圖3(b)的Cu LMM俄歇譜可以觀察到,圖中有兩個峰在 918.7和916.4 eV,分別歸屬于單質Cu和Cu2O。并且在918.7 eV處的峰很突出,916.4 eV處出現的峰很微弱,這表示在TiO2納米管表面鏤空區沉積的主要是單質Cu,以及微量的Cu2O。
3.2 光誘導約束刻蝕后TiO2納米管表面Cu的沉積機制
關于Cu2+在TiO2表面光催化還原的生成機制和產物組成存在著不同的報道。Bideau等10認為,在TiO2上光催化還原沉積的產物為Cu(I)和Cu(0)的混合物。Foster等11認為,在光還原過程中,TiO2表面紫色的Cu沉積為Cu(I),是由Cu2+被TiO2生成的光生電子還原而成的。Lv等6認為,Cu2+是直接被光催化還原而成Cu(0),產物主要是Cu(0)的納米粒子。這些不同可能是由于不同的反應條件引起的,如TiO2的晶型結構、形貌及摻雜,Cu2+的濃度,光照的強度和光照的時間等等,都會影響沉積及組成12。因此有必要研究光誘導約束刻蝕中TiO2納米管表面Cu的沉積機制。

圖2 光誘導約束刻蝕Cu的TiO2 NTs表面的SEM及EDS圖Fig.2 SEM and EDS images of TiO2 NTs after photoinduced confined etching.(a, c) before etching, (b, d) after etching. 1% (w) glycine solution; pH ~5.8.

圖3 光誘導約束刻蝕后TiO2納米管表面的XPS能譜圖Fig.3 Surface XPS spectra of TiO2 NTs after photoinduced confined etching.(a) Cu 2p, (b) Cu LMM.
光誘導約束刻蝕 Cu主要包括三個化學反應過程和三個傳質過程。三個化學反應過程分別是光誘導產生刻蝕劑?OH、?OH 被清除、和?OH 與Cu發生氧化反應。三個傳質過程分別是刻蝕劑?OH的傳質過程、Cu被氧化后的產物Cu(II)的傳質過程、和約束劑甘氨酸的傳質過程。實驗過程中未攪拌,可忽略對流的作用。在工具TiO2納米管和工件Cu上都未施加電壓,電遷移作用也可忽略。所以傳質過程主要以擴散的方式進行,即主要是刻蝕劑?OH從TiO2納米管表面到Cu表面的擴散,氧化之后的Cu(II)從工件Cu表面向溶液本體的擴散,及溶液中的甘氨酸向TiO2納米管與Cu之間的液層擴散。當這個液層縮小到微納尺度時,三種擴散都受到很大限制。圖 4(a)是光誘導約束刻蝕過程中刻蝕以及光催化沉積 Cu的機理示意圖。TiO2納米管在光激發下將同時產生空穴和電子,前者輸運到表面構成氧化位點,而后者輸運到表面產生還原位點。一方面,空穴在溶液環境中氧化生成刻蝕劑?OH,?OH擴散到Cu表面并將其刻蝕成 Cu2+,并與溶液中的約束劑甘氨酸(gly)形成可溶性絡合物[Cu(gly)2]2+,該絡合物將脫離表面從而實現工件 Cu表面的連續刻蝕。另一方面,光生電子傾向于沿納米管管軸向傳輸,既會沿著納米管傳輸到電極基底,也可能傳輸到納米管頂端13,隨著光照時間增加,在納米管頂端的光生電子有可能發生一定程度的富集。由于TiO2的光生電子導帶電位(CB)為?0.53 V (vs NHE (標準氫電極))14,E0(Cu2+/Cu0) = +0.340 V (vs NHE)15,當添加絡合劑甘氨酸后,根據 Cu2+和甘氨酸及 Cu+和甘氨酸的二級絡合常數lgβ,分別為15和10,計算得到E0([Cu(gly)2]2+/Cu0)、E0([Cu(gly)2]+/Cu0)和 E0([Cu(gly)2]2+/[Cu(gly)2]+)分 別 為 ?0.103、?0.0712 和?0.138 V (vs NHE),均高于 TiO2的光生電子的電位16。因此,從熱力學上說Cu2+被TiO2的光生電子還原是可行的。由于工具–工件之間的距離很小,為微米數量級,[Cu(gly)2]2+向外的擴散受到限制,隨著光照時間的增加及刻蝕反應的進行,刻蝕產物[Cu(gly)2]2+將在該微米級別的液層之間逐漸累積,并且擴散到工具TiO2納米管表面,從 TiO2納米管上得到電子還原生成 Cu(0),生成的 Cu(0)還會進一步被溶液中含有的微量氧氣氧化6。不同的是,跟納米微粒膜相比,由于光生電子在納米管末端沉積的 Cu顆粒上的富集使其較不容易被進一步氧化成 Cu2O,從而生成的Cu2O量較少。因此,在光誘導約束刻蝕體系中沉積在 TiO2納米管表面的沉積產物主要為單質Cu(0),以及隨后氧化生成的微量Cu2O。

圖4 (a)光誘導約束刻蝕體系中Cu的刻蝕和光催化沉積的機制示意圖和(b) Cu2O, TiO2和Cu的能帶及費米能級圖,pH = 7Fig.4 (a) Mechanism of the etching and photodeposition of Cu in photoinduced confined etching system (b) Energy band and Fermi level of Cu2O, TiO2 and Cu, pH = 7.
由于光誘導約束刻蝕反應后TiO2納米管表面只有局部區域有Cu沉積,且沉積量有限,用常規的光電化學表征方法難以精確評估不同參數條件下沉積在TiO2納米管上的Cu對于光電轉換效率的影響。因此我們采用Cu(NO3)2和甘氨酸的混合溶液來模擬研究Cu2+在TiO2納米管表面的光催化還原沉積及對TiO2納米管光催化性能影響。通過比較不同光還原時間下 TiO2納米管表面的 SEM圖(圖5(a–c))可看出,TiO2納米管表面先是出現一些Cu的小顆粒,隨著光還原時間的增加,Cu顆粒尺寸逐漸變大,表面的覆蓋度也增大。這是由于,當少量Cu在表面成核后,在Cu和TiO2納米管的接觸面上,由于Cu的費米能級低于TiO2導帶的費米能級,如 4(b)所示,導帶上的電子會向金屬顆粒遷移直至二者表面的費米能級相等17。因而隨著反應的進行,最初生成的金屬Cu顆粒會因聚集導帶上的電子而作為后續還原沉積反應的形核核心,而起到催化的作用,使得Cu顆粒不斷長大。
模擬液的pH為2.5時,光還原得到的Cu顆粒分布較寬易出現大尺寸顆粒且帶有整齊的表面和棱角。Foster等18指出溶液的 pH在 0.60?6.60范圍內,pH對Cu2+的光催化還原有著重要的影響。在堿性條件下,Cu離子易水解生成沉淀(光誘導約束刻蝕體系中生成的Cu離子濃度較低,不易出現此問題),因此我們進一步在pH為5.8的環境下考察模擬液中光催化沉積 Cu的形貌。如圖 5(d)的SEM圖所示,光催化還原60 min后,TiO2納米管表面沉積的Cu與pH 2.5下得到的Cu相比,更像是由許多小顆粒團聚而成的疏松的團簇,與光誘導約束刻蝕 Cu體系中的沉積 Cu形貌更為相近。但因為該模擬溶液中的 Cu離子濃度遠高于刻蝕體系的,因此沉積反應的速度快得多,使沉積的Cu顆粒更為小和分散。
從Cu/TiO2NTs復合電極表面的XPS譜(圖S1(Supporting Information))可知,Cu 2p3/2和Cu 2p1/2分別為932.2和952.0 eV,與單質Cu相符,而且沒有出現特征的CuO的震激伴峰,說明電極表面不存在CuO。從圖S1(b)的Cu LMM俄歇譜來可觀察到,圖中有兩個峰分別歸屬于單質Cu和Cu2O。這表明模擬液中在TiO2納米管表面沉積的是單質Cu和Cu2O的混合物,和光誘導約束刻蝕體系中的成分的結果類似,但模擬液中沉積產物中Cu2O的含量較高。可能原因是該 Cu2+濃度下顆粒生長較快,難以生成大顆粒且顆粒間堆積較為疏松,存在大量的晶界,使得光生電子在Cu顆粒間傳輸時容易被捕獲,失去對Cu的陰極保護,從而較為容易被氧化成Cu2O。

圖5 不同光還原時間下Cu/TiO2 NTs電極的SEM圖Fig.5 SEM of photo-reduced Cu/TiO2 NTs under different irradiation times.(a) 10 min; (b) 30 min; (c) 60 min; (d) 60 min; Solution contained 1% (w) glycine and 1.25% (w) Cu(NO3)2; (a, b, c) pH ~2.5; (d) pH ~5.8.

圖6 TiO2 NTs和Cu/TiO2 NTs電極的吸收光譜圖Fig.6 Absorption spectra of TiO2 NTs and Cu/TiO2 NTs.
為考察沉積Cu對TiO2納米管光催化性能的影響,分別測定了 TiO2納米管陣列光電極和Cu/TiO2納米管陣列復合電極的吸收光譜和暫態光電流響應。圖 6是采用固體透射方式測定的Cu/TiO2納米管陣列復合電極的紫外可見吸收光譜。由圖可見,沉積Cu后,復合電極的吸收帶邊由380 nm左右拓展到了500 nm左右,且350 nm以后的吸收均有明顯增加。圖 7是在白光照射下外加電位為0 V時的TiO2納米管陣列電極和不同光催化還原時間下制備的 Cu/TiO2納米管陣列電極的暫態光電流響應。從圖7(a)的TiO2納米管陣列電極的陽極光電流響應曲線中,未觀察到光照開始和停止瞬間由表面態引起的明顯的前后峰,由此可見所制備的TiO2納米管晶型較好、表面態濃度小。從圖 7(b)中三種光催化還原時間制備的Cu/TiO2納米管復合電極的暫態光電流響應曲線可見,復合電極的光電流響應明顯強于TiO2納米管電極的,且隨著光催化還原時間的增加,所得到的復合電極的光電流響應逐漸增強。與TiO2納米管電極不同的是:在光照開始時,復合電極會出現一個先增大后減小的光電流瞬變。這可歸咎于沉積Cu后TiO2納米管陣列電極表面缺陷態增加。在光照瞬間,光生電子和空穴快速分離,光生電子的電化學復合與光生電子朝體相遷移到外電路產生一個正向的陽極光電流;由于表面缺陷態的存在,光生電子和空穴隨即也會掉進表面態能級,發生表面態復合,使陽極光電流減小。之后,隨著光生電子和空穴的分離及復合的動力學過程逐步趨于平衡,光電流又趨于穩定19。當沉積時間增加,復合電極的光電流增大。這首先可歸咎于光吸收的增加(由吸收光譜圖可見);其次,電子在TiO2和Cu之間重新富集,并由TiO2注入Cu中17,Cu作為電子捕獲劑,阻止空穴和電子的重組。并且,激發電子快速轉移到Cu上會增強電子和空穴的分離,因此光電流將明顯增加9。第三,根據文獻報道,適量的Cu2O摻雜不僅含有豐富的電子陷阱,也會捕獲光生電子,從而有效地抑制光生載流子的復合,提高TiO2的光催化活性20。然而當光催化還原時間較長,如 60 min時,Cu/TiO2納米管復合電極光電流響應又發生較大的變化:光電流先出現一個表面態的尖峰后,不是逐步趨向穩態而是又緩慢升高。我們認為,這可能是由于長時間沉積的Cu和Cu2O數量多到一定程度,作為p-型半導體的Cu2O自身的光電轉換效應已不可忽略:剛開始其光生電子被溶液捕捉產生的陰極光電流成份會抵消一部分TiO2產生的陽極光電流;隨著光照持續進行,由于能帶匹配(見圖4b的能帶圖)14,21,Cu2O導帶上富集的電子又會通過Cu2O和TiO2之間的異質結界面傳輸給TiO2導帶,反而增強TiO2的陽極光電流,隨著這兩個過程慢慢達到平衡,光電流也慢慢升高達到穩態。雖然實驗結果表明,在目前條件下工具表面TiO2上沉積Cu有利于提高光催化性能,但對于光誘導約束平坦化的應用而言,也存在兩個潛在的不利因素:第一,過量的Cu沉積可能會造成會破壞納米管的原有形貌,造成工具表面的不平整,不利于平坦化;第二,反應過程中不斷增加的沉積 Cu的光催化性能不利于控制刻蝕的速度和平坦化精度。因此,接下來我們也繼續探索如何控制光誘導約束刻蝕的實驗條件,以抑制納米管表面Cu的沉積。
3.4 TiO2納米管表面Cu沉積的抑制
刻蝕產物在工具–工件之間狹小空間內的擴散受限是TiO2納米管上Cu沉積的主要原因之一。因此,抑制Cu沉積的思路之一就是改善刻蝕產物在工具–工件之間的傳質。我們同時也嘗試了通過引入攪拌和添加絡合劑來控制工具表面TiO2層上的Cu沉積。
3.4.1 Cu沉積抑制實驗中的pH的選擇
pH既會影響TiO2的光催化,又會影響Cu離子的光催化還原。我們上一節在模擬液中的實驗結果表明,在pH 5.8下Cu沉積相對于2.5下要快。Kabra等22指出溶液的 pH在2?10范圍內,Cu2+的光催化沉積的量在pH為7左右達到最大值。而我們課題組之前的研究發現,pH為10左右時生成的游離態?OH 濃度最高23。因此,我們比較了pH為5.8和10時TiO2納米管表面Cu的沉積情況及Cu表面的刻蝕情況。
分別對在pH 5.8下和pH 10光誘導約束刻蝕后的Cu表面和TiO2納米管表面做SEM,結果如圖S2 (Supporting Information)所示。比較圖S2(a)和(b)的TiO2表面可見,pH 10下TiO2納米管表面沉積的Cu比pH 5.8下的顆粒更小且更為分散和均勻。這是因為pH 10下產生的·OH濃度更高,更容易刻蝕Cu表面,產生更多的Cu2+,使沉積的速度更快,因此沉積的面更廣,顆粒更小。比較S2(c)和(d)的Cu表面可見,pH 10下Cu表面的刻蝕比pH 5.8下Cu表面的刻蝕更明顯,這與pH 10下產生的·OH濃度更高的結論是相一致的。因此,后續選用溶液pH為10用于抑制Cu沉積的光誘導約束刻蝕實驗。一來是因為刻蝕的效果更明顯;二來是因為沉積的Cu的量足夠大,更方便進行檢測和比較。
3.4.2 引入攪拌和加入絡合劑
引入外置攪拌,將在一定程度上促進工具–工件之間的液層與本體溶液的對流傳質,使液層中的刻蝕產物[Cu(gly)2]2+盡快地被帶離,降低其濃度,并使約束劑得到不斷的補充,從而有效地增加Cu表面的刻蝕進程。為了探究引入攪拌對光誘導約束刻蝕體系的影響和抑制TiO2納米管表面沉積Cu的情況,在pH 10下引入一系列轉速,并進行光誘導約束刻蝕。其中不加轉速、引入 1000 r?min?1轉速的光誘導約束刻蝕后的TiO2納米管表面和 Cu表面的 SEM 表征分別如圖 8和圖 S3(Supporting Information)所示。1000 r?min?1的圖 8(b)相比于0 r?min?1的圖8(a),沉積的Cu顆粒更大。這是由于當引入1000 r?min?1的外置轉速后,外界對流在一定程度上能使液層中的[Cu(gly)2]2+更快地被帶離,并補充約束劑,因此液層中的[Cu(gly)2]2+濃度更低,使得 Cu的生長更慢。但TiO2和Cu之間的距離相對自身的尺寸較小,隨著時間的推移,刻蝕產物仍然將持續積累在距離很小的液層中,因此仍存在Cu的沉積。比較圖S3(a)和 S3(b)可見,當引入 1000 r?min?1的外置轉速后,Cu表面的刻蝕情況有所加劇。這是因為外置攪拌會使液層中的[Cu(gly)2]2+更快地被帶離,使刻蝕反應向正向移動,因此Cu表面的刻蝕情況隨轉速增加有所提高。由此可見,加入外置攪拌會抑制Cu的沉積,同時加劇刻蝕的程度。
在光誘導約束刻蝕體系中,絡合劑的加入會增加Cu的去除率,防止形成氧化物膜進一步氧化。但同時絡合劑還可能被?OH氧化作為約束劑,起到約束劑的作用。因此,加入一個能絡合Cu且不約束?OH的絡合劑將有助于對Cu表面的約束刻蝕。為了比較不同的絡合劑對TiO2納米管產生?OH的影響,分別做了不同絡合劑對游離?OH濃度影響的熒光檢測,如圖S4 (Supporting Information)所示。當加入尿素后,對?OH影響較小。對于氨水,熒光峰(曲線 c)很低,說明氨水會約束?OH24。至于EDTA,熒光峰(曲線d)很低,這是因為EDTA會作為空穴捕獲劑25,使得?OH 的產生量下降。因此,只有尿素(曲線b)對體系中游離?OH影響最小,而其他的絡合劑均會與?OH 反應或者抑制?OH 的生成,因此這里選用尿素作為引入的絡合劑。
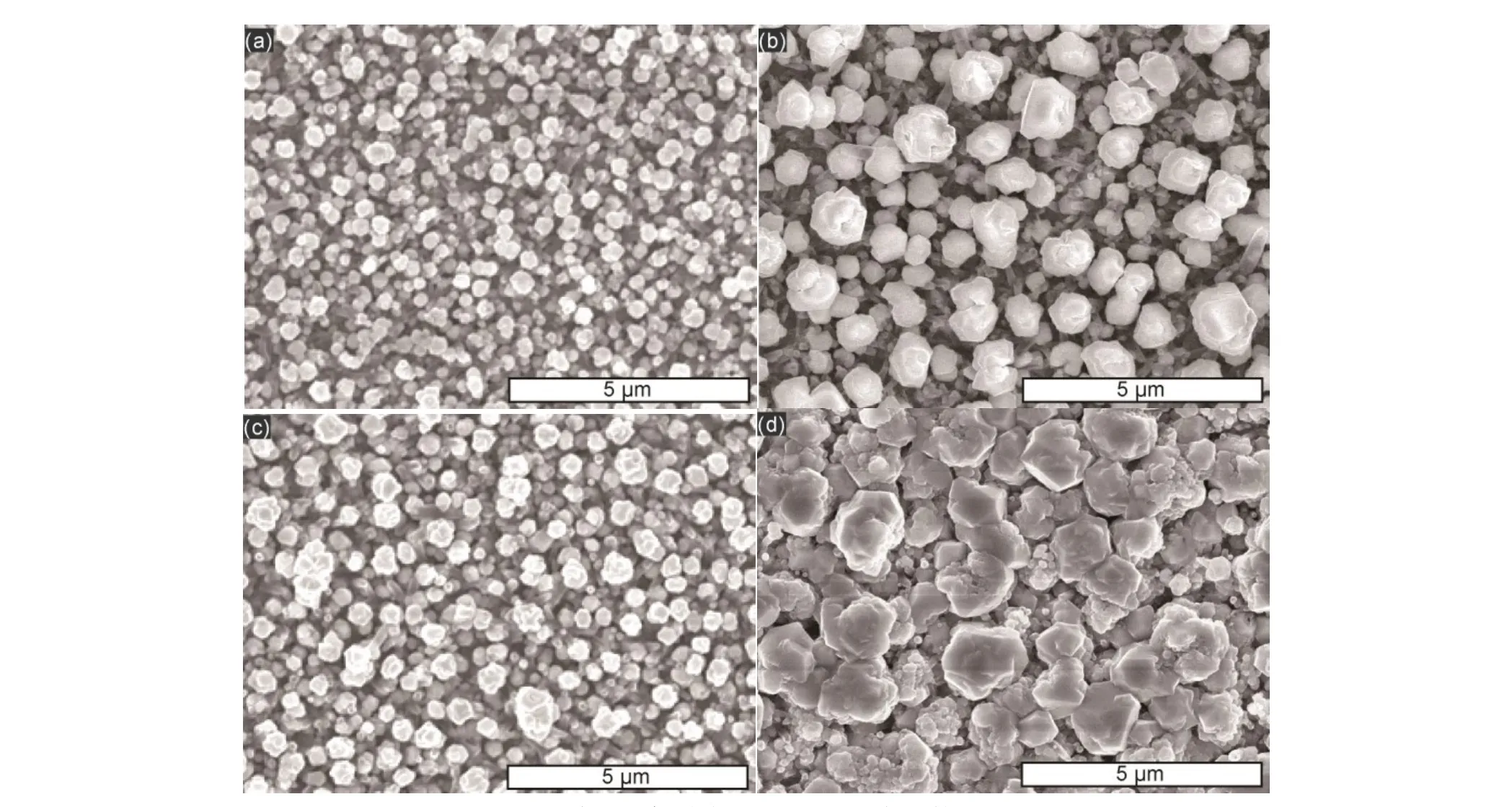
圖8 光誘導約束刻蝕后TiO2 NTs表面的SEM圖Fig.8 SEM of TiO2 NTs after photoinduced confined etching.(a) 0 r?min?1, 1% (w) glycine; (b) 1000 r?min?1, 1% (w) glycine; (c) 0 r?min?1, 1% (w) glycine + 0.05 mol·L?1 urea;(d) 1000 r?min?1, 1% (w) glycine + 0.05 mol·L?1 urea.
為了比較加入絡合劑對光誘導約束刻蝕體系的影響和抑制TiO2納米管表面沉積Cu的情況,分別對在pH 10下未攪拌、引入1000 r?min?1攪拌的光誘導約束刻蝕及加入尿素后未攪拌和 1000 r?min?1攪拌的光誘導約束刻蝕后,Cu表面和TiO2納米管表面做SEM。比較未攪拌的圖8(a, (c),及比較 1000 r?min?1攪拌的圖 8(b, d)可見,當加入絡合劑后,TiO2納米管表面沉積的Cu的顆粒更大,這和引入外置攪拌的原因一致,同樣是因為Cu的生長速度更慢,使得顆粒更大,Cu的沉積受到了抑制。因此,加入外置攪拌和加入絡合劑會更有利于Cu沉積的抑制。而加入尿素和1000 r?min?1攪拌的圖 8(d)比未攪拌和加尿素的圖 8(b)沉積顆粒更大,也是因為沉積的Cu生長速度更慢,沉積受到了抑制。對于刻蝕后的Cu表面,比較未攪拌的圖 S3(a)和 S3(c),及比較加入 1000 r?min?1的圖S3(b)和 S3(d)可見,當加入尿素后,Cu表面的晶型重整程度都有所減少,并且表面趨向于更加平坦。這是由于當 Cu表面與高濃度·OH反應,Cu氧化速度雖然較快,但是伴隨著Cu的原位沉積26,長時間后Cu表面將出現晶型重整。當加入絡合劑后,加入的絡合劑將提高刻蝕產物被絡合的程度,減少Cu的原位沉積。而減少刻蝕過程的晶型重整將有利于提高平坦化的程度。該結果在圖S3(d)中最為明顯。因此,加入外置攪拌的同時,加入絡合劑不僅會提高約束刻蝕的程度,也將可能更有利于表面的平坦化。
由以上結果可見,通過改善傳質來抑制Cu的沉積的方法雖然會在一定程度上增強光誘導約束刻蝕的程度,但未必都有利于Cu的表面的平坦化。由于工具和工件間的微納米尺度液層中涉及多個化學和傳質過程,如引入攪拌在改變刻蝕產物的傳質的同時也會改變刻蝕劑的傳質過程,使刻蝕劑在液層之間的分布變得不均一,即約束刻蝕層厚度的不均一,從而影響工件的拋光效果。為了解決這個問題,可在引入攪拌的同時通過控制工具和工件之間的相對運動(如相對旋轉),將能有效地使拋光更均一。因此在抑制Cu沉積的方法和條件參數的選擇上需同時評估多方面的影響。
4 結 論
通過SEM、EDS及XPS表征發現,在光誘導約束刻蝕Cu的過程中,工具TiO2納米管表面會沉積單質Cu及少量Cu2O。Cu的沉積是由工具–工件之間累積的約束刻蝕產物[Cu(gly)2]2+在 TiO2納米管表面被光激發下產生的電子還原生成的,而微量的Cu2O是由單質Cu隨后氧化生成的。通過采用 Cu(NO3)2和甘氨酸的混合溶液模擬研究Cu2+在 TiO2納米管表面的光催化還原沉積及對TiO2納米管光催化性能的影響,進一步解釋了沉積機制并發現沉積增強了TiO2納米管的光催化性能:當少量Cu在表面成核后,會因聚集導帶上的電子而作為后續還原沉積反應的形核核心,使得Cu顆粒不斷長大;當TiO2納米管表面沉積上Cu后,Cu/TiO2復合電極的吸收帶邊由380 nm左右拓展到了500 nm左右,增強了光吸收;隨著光催化還原時間的增加,復合電極的光電流逐漸增強;還發現當光催化還原時間過長時,Cu2O自身的光電轉換效應已不可忽略,出現不同的暫態光電流響應。雖然沉積Cu有利于增強TiO2納米管的光催化性能,但過量的Cu沉積可能會改變工具表面的原有形貌,且反應過程中不斷增加的沉積Cu不利于控制刻蝕速度和平坦化精度。初步的抑制實驗表明:加入外置攪拌會使Cu的沉積更慢,在一定程度上抑制Cu的沉積,但加劇刻蝕的程度;而攪拌的同時加入絡合劑尿素可能會更有利于 Cu表面的平坦化。所以,抑制Cu的沉積需要兼顧提高約束刻蝕和平坦化程度,抑制的方法和條件的選擇需要同時考慮在微納尺寸的工具–工件之間液層的多種化學反應和傳質過程。
Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Tian, Z. W.; Fen, Z. D.; Tian, Z. Q.; Zhuo, X. D.; Mu, J. Q.; Li, C. Z.;Lin, H. S.; Ren, B.; Xie, Z. X.; Hu, W. L. Faraday Discussions 1992,94, 37. doi: 10.1039/FD9929400037
(2) Zhang, L.; Ma, X. Z.; Lin, M. X.; Lin, Y.; Cao, G. H.; Tang, J.; Tian,Z. W. J. Phys. Chem. B 2006, 110, 18432. doi: 10.1021/jp063110m
(3) Zu, Y. B.; Xie, L.; Mao, B. W.; Tian, Z. W. Electrochim. Acta 1998,43, 1683. doi: 10.1016/S0013-4686(97)00301-0
(4) Fang, Q. Y.; Zhou, J. Z.; Zhan, D. P.; Shi, K.; Tian, Z. W.; Tian, Z. Q.Chem. Commun. 2013, 49, 6451. doi: 10.1039/c3cc42368a
(5) Montini, T.; Gombac, V.; Sordelli, L.; Delgado, J. J.; Chen, X.;Adami, G.; Fornasiero, P. ChemCatChem 2011, 3, 574.doi: 10.1002/cctc.201000289
(6) Lv, X. J.; Zhou, S. X.; Zhang, C.; Chang, H. X.; Chen, Y.; Fu, W. F.J. Mater. Chem. 2012, 22, 18542. doi: 10.1039/c2jm33325b
(7) Canterino, M.; Somma, I. D.; Marotta, R.; Andreozzi, R. Water Res.2008, 42, 4498. doi: 10.1016/j.watres.2008.07.035
(8) Wu, N. L.; Lee, M. S. Int. J. Hydrog. Energy 2004, 29, 160.doi: 10.1016/j.ijhydene.2004.02.013
(9) Tseng, I. H.; Chang, W. C.; Wu, J. C. S. Appl Catal B:Environ 2002, 37, 37. doi: 10.1016/S0926-3373(01)00322-8
(10) Bideau, M.; Claudel, B.; Faure, L.; Rachimoellah, M. Chem.Eng. Commun. 1990, 93, 167.doi: 10.1080/00986449008911444
(11) Foster, N. S. ; Noble, R. D. ; and Kovel, C. A. Environ. Sci.Technol. 1993, 34, 3865. doi: 10.1021/es00039a016
(12) Jacobs, J. W. M.; Kampers, F. W. H.; Rikken, J. M. G.;Bulle-Lieuwma, C. W. T.; Koningsberger, D. C.J. Electrochem. Soc. 1989, 136, 2914.doi: 10.1149/1.2096373
(13) Beranek, R.; Tsuchiya, H.; Sugishima, T.; Macak, J. M.;Taveira, L.; Fujimoto, S.; Kisch, H.; Schmuki, P. Appl. Phys.Lett. 2005, 87, 167. doi: 10.1063/1.2140085
(14) Xiong, Z. G.; Zhao, X. S. J. Mater. Chem. A 2013, 1, 7738.doi: 10.1039/c3ta11247k
(15) Tavares, M. C.; Machado, S. A. S.; Mazo, L. H. Electrochim.Acta 2001, 46, 4359. doi: 10.1016/S0013-4686(01)00726-5
(16) Kiss, T.; Sovago, I.; Gergely, A. Pure Appl. Chem., 1991, 63,597. doi: 10.1351/pac199163040597
(17) Wood, A.; Giersig, M.; Mulvaney, P. J. Phys. Chem. B 2001,105, 8810. doi: 10.1021/jp011576t
(18) Foster, N. S.; Lancaster, A. N.; Noble, R. D.; Koval, C. A. Ind.Eng. Chem. Res. 1995, 34, 3865. doi: 10.1021/ie00038a025
(19) Tafalla, D.; Salvador, P.; Benito, R. M. J. Electrochem. Soc.1990, 137, 1810. doi: 10.1149/1.2086809
(20) Xin, B. F.; Wang, P.; Ding, D. D.; Liu, J.; Ren, Z. Y.; Fu, H. G.Appl. Surf. Sci. 2008, 254, 2569.doi: 10.1016/j.apsusc.2007.09.002
(21) Bessekhouad, Y.; Robert, D.; Weber, J. V. Catal. Today 2005,101, 315. doi: 10.1016/j.cattod.2005.03.038
(22) Kabra, K.; Chaudhary, R.; Sawhney, R. L. J. Hazard. Mater.2007, 149, 680. doi: 10.1016/j.jhazmat.2007.04.028
(23) Hu, Y.; Fang. Q. Y.; Zhou, J. Z.; Zhan, D. P.; Shi, K.;Tian, Z.Q.; Tian, Z. W. Acta Phys. -Chim. Sin., 2013, 29 (11) , 2392.[胡 艷, 方秋艷, 周劍章, 詹東平, 時 康, 田中群, 田昭武. 物理化學學報, 2013, 29 (11) , 2392.]doi: 10.3866/PKU.WHXB201309043
(24) Hickel, B.; Sehested, K. Radiat. Phys. Chem. 1992, 39, 355.doi: 10.1016/1359-0197(92)90244-A
(25) Qu, P.; Zhao, J. C.; Shen, T.; Hidaka, H. J. Mol. Catal. A:Chem. 1998, 129, 257. doi: 10.1016/S1381-1169(97)00185-4
(26) Kirino, O.; Enomoto, T. Precis. Eng. 2011, 35, 669.doi: 10.1016/j.precisioneng.2011.05.0
Deposition and Inhibition of Cu on TiO2Nanotube Photoelectrode in Photoinduced Confined Etching System
HUANG Ya-Yu FANG Qiu-Yan ZHOU Jian-Zhang*ZHAN Dong-Ping SHI Kang TIAN Zhong-Qun
(State Key Laboratory of Physical Chemistry of Solid Surfaces and Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, Fujian Province, P. R. China)
A photoinduced confined etching system was used for the unstressed chemical planarization of Cu. Cu deposits were found on the surface of TiO2nanotubes of the tool during the photoinduced confined etching of the Cu workpiece. Scanning electron microscopy, energy-dispersive X-ray spectroscopy, and X-ray photoelectron spectroscopy were used to analyze the morphology and composition of the Cu deposits, and the mechanism of the photodeposition of Cu in the micro/nanoscale liquid layer between the tool and the workpiece was investigated. Moreover, a simulated cupric solution was used to study the effect of the Cu deposits during the photoinduced confined etching. Several routes including stirring and complexing agent were used to investigate the inhibition of Cu deposition on the surface of TiO2nanotubes and the simultaneous effect on the etching of Cu workpiece. The results showed that the Cu deposits enhanced the photocatalytic performance of TiO2nanotubes, but the mechanism of enhancement changed with the increase in Cu deposits. Inhibition of Cu deposition byimproving mass transfer can lead to the increase in the etching of Cu; addition of complexing agent combined with enhanced mass -transfer can inhibit Cu deposition, while improving the planing effect.Thus, the choice of inhibition methods and conditions should balance the effect of the micro/nano liquid layer between the tool and workpiece on multiple chemical reactions and mass transfer processes. The results provide an important guiding significance for further regulation and optimization of the photoinduced confined etching system.
Photo-induced confined etching; TiO2nanotube arrays; ?OH; Scavenging agent;Photo-deposition of Cu; Mass transfer
April 13, 2017; Revised: May 2, 2017; Published online: May 12, 2017.
O649
10.3866/PKU.WHXB201705125 www.whxb.pku.edu.cn
*Corresponding author. Email: jzzhou@xmu.edu.cn; Tel: +86-0592-2189663.
The project was supported by the National Natural Science Foundation of China (91023043, 21021002, 91023006).
國家自然科學基金(21273182, 21533006, 21621091)資助項目
? Editorial office of Acta Physico-Chimica Sinica
