鈰基復合氧化物催化劑在SiO2表面的失活機制
2017-11-01 17:29:18占林軍孫曉燕朱秋蓮陳銀飛盧晗鋒
物理化學學報 2017年7期
關鍵詞:催化劑
占林軍 孫曉燕 周 瑛 朱秋蓮 陳銀飛 盧晗鋒
(浙江工業大學化學工程學院,杭州 310014)
鈰基復合氧化物催化劑在SiO2表面的失活機制
占林軍 孫曉燕 周 瑛*朱秋蓮 陳銀飛 盧晗鋒
(浙江工業大學化學工程學院,杭州 310014)
以甲苯催化燃燒為模型反應,通過調節不同Cu-Mn-Ce (CMC)復合氧化物在多孔SiO2(KIT-6)上的負載量,研究了SiO2表面與CMC作用對催化劑物理化學性能的影響。發現低負載量下CMC氧化物出現明顯失活現象,與SiO2接觸會抑制氧化物活性相的形成,SiO2量的減少可使CMC復合氧化物活性得到逐步恢復。X射線衍射(XRD)、程序升溫還原(H2-TPR)、N2吸附(BET)和透射電鏡(HRTEM)等表征表明,SiO2不對CMC晶相結構產生影響,這種失活機制是由于SiO2表面的豐富羥基作用,導致表面氧化物高度分散,活性氧物種從晶格氧轉變為表面氧。復合氧化物的晶格氧對催化燃燒起到關鍵性作用,通過焙燒去除SiO2表面羥基和減少SiO2用量,可使復合氧化物晶格氧的數量增加,恢復復合氧化物催化劑活性。
SiO2;Cu-Mn-Ce復合氧化物;失活;載體效應;催化燃燒
1 引 言
在氧化物材料中,SiO2可構造性最強,通過自組裝技術,可以構建不同規整孔道結構的介孔材料1?3,如 MCM-414?7,SBA-158?10,KIT-611?13等。這些材料的合成為新型催化劑發展提供了強有力的推動力。尤其是一些難以合成介孔結構的活性金屬氧化物材料14,可以以這些介孔SiO2作為硬模板劑,通過納米鑄造(Nanocasting)技術來合成高比表面積的規整的氧化物介孔材料,從而進一步提高比表面積和催化活性位的分散,并為反應物和產物分子提供擴散通道,提高氧化物催化劑的催化性能15?17。這個技術雖然得到相當廣泛的應用,但也發現,在以SiO2為模板構筑過程中,需要把 SiO2模板通過堿液或 HF洗滌掉18,19。但多次洗滌總是會有SiO2殘留20,21,這些殘留的SiO2會對活性金屬氧化物物理化學特性產生怎樣的影響?目前還沒有系統的研究。
另外許多文獻認為,SiO2是相對較為惰性的載體,以高比表面積的介孔SiO2作為活性氧化物催化劑載體,使活性金屬氧化物在表面充分暴露,是目前新型氧化物催化劑構筑方面的一個熱點研究方向22?24。但我們前期研究發現,SiO2雖然惰性,但也會對表面活性氧化物組分結構產生特定的影響,尤其是鈰基復合氧化物催化劑,在極低的負載量情況下,鈰基氧化物催化劑會出現明顯的失活現象25。而這種失活現象是怎樣產生的,SiO2和鈰基氧化物之間的作用機制如何?都需要進一步研究和探索。
而論文正是基于此,通過浸漬法制備了以KIT-6為載體的負載型 Cu-Mn-Ce復合氧化物(縮寫為 CMC)催化劑并用堿液多次洗滌去除 SiO2,以甲苯催化燃燒為模型反應,考察SiO2對鈰基復合氧化物催化特性影響,并利用 XRD,BET,HRTEM,H2-TPR等表征手段揭示SiO2和鈰基氧化物之間的失活作用機制。
2 實驗部分
2.1 催化劑的制備
將6 g P123溶解在217 g蒸餾水中,并加入11.8 g的36%的鹽酸。在35 °C下,向溶液中加入6 g正丁醇,連續攪拌1 h;加入12.9 g正硅酸乙酯,在35 °C下攪拌24 h。將得到的混合溶液轉入晶化釜中,在100 °C下水熱晶化24 h后得到白色懸浮液,過濾,在100 °C 下干燥12 h,然后在550 °C下焙燒3 h,得到KIT-6介孔分子篩26,27。
負載型 Cu-Mn-Ce/KIT-6 (SiO2載體質量分數量分別為 79%,65%,34%)催化劑的制備采用浸漬法制備。以Cu、Mn和Ce的硝酸鹽為原料,配制Cu/Mn/Ce物質的量之比為1 : 2 : 4混合溶液,再加入與總金屬離子等物質量的檸檬酸,攪拌溶解成均一溶液。然后向溶液中加入一定量的KIT-6載體,室溫攪拌12 h,在真空旋轉蒸發儀中揮發水分,在100 °C下干燥12 h、研磨、500 °C焙燒3 h制得催化劑,標記為CMC/w%-KIT-6,w為載體量(%)。
SiO2的去除在70 °C,濃度為2 mol·L?1的NaOH溶液中進行,將制得的CMC/w%-KIT-6催化劑浸入NaOH溶液中攪拌洗滌N次(N = 1、2、3、4、5),每次洗滌30 min,去除不同質量的SiO2,之后過濾、去離子水洗滌3次、100 °C下干燥12 h后得到的催化劑,標記為 CMC/w%K-N,N為堿洗次數。
2.2 催化劑的表征
樣品的晶相結構在荷蘭 PNAlytical公司 X? Pert Pro型X射線衍射儀(Ni濾波,Cu Kα輻射源)上測定,管電壓為45 kV,管電流為40 mA,掃描范圍 2θ = 10°?80°,步長為 0.02 (°)·s?1。
催化劑的織構性質(比表面積、孔體積和吸附脫附等溫線)的測定采用低溫(?196 °C)氮氣吸附法,在Micromeritics公司的3Flex型表面性質分析儀測試。吸附測定前,樣品先在250 °C脫氣預處理6 h。
H2-TPR實驗在衢州泛泰生產的FINESORB-3010E型化學吸附儀上進行。活性組分裝填量為0.2 g,CuO作為標樣。首先在Ar氣氛下200 °C預處理1 h,冷卻至100 °C,然后通入5% H2/Ar的還原氣,催化劑在100 °C下吹掃20 min,再以 10 °C·min?1的速率升溫至 750 °C,最后又通過Ar氣吹掃降溫,其中載氣流速均為30 mL·min?1。熱導檢測器(TCD)檢測,TCD 池溫度為60 °C,TCD電流為60 mA。
樣品的微觀形貌采用荷蘭Philips-FEI公司生產的Tecnai G2 F30 S-Twin 300 kV高分辨率透射電子顯微鏡觀察,加速電壓為30 kV。
2.3 催化劑的活性評價
催化劑活性測試在常壓連續流動氣固相反應裝置上進行,石英反應管內徑為6 mm,按照計算催化劑活性組分用量0.3 g,甲苯氣體發生器置于冰水物(0 °C)中,甲苯進料濃度控制在 3000 mg·m?3。反應空速為 24000 mL·g?1·h?1。反應氣氛為甲苯-空氣混合氣。反應尾氣采用捷島 GC1620色譜儀六通閥直接進樣在線檢測出口有機物濃度,FID檢測器檢測出口有機物濃度。
3 結果與討論
3.1 SiO2對CMC復合氧化物活性影響
圖1(a)是以KIT-6介孔分子篩為載體的CMC催化燃燒甲苯活性圖,圖中催化劑在測試時保持活性物質CMC反應空速不變(以CMC為重量基準來計算空速)。發現隨著載體KIT-6用量增加,其活性出現顯著下降,當載體量(SiO2含量)為 79%時,催化劑完全失活。這是一個非常有趣的實驗現象。為進一步考察SiO2量的影響,通過NaOH堿洗方式,把三個 CMC/SiO2中SiO2溶解去除,每個催化劑都在2 mol·L?1的NaOH溶液中洗滌3次,得到的催化劑再次進行活性測試(見圖 1(b)),發現當把SiO2去除后,其活性都得到了恢復。圖1(c, d)中測試了 CMC/65%KIT-6催化劑堿洗次數和活性之間的關聯,隨著堿洗次數增加SiO2量的減少,催化劑活性得到逐步恢復,到第4、5次基本去除SiO2下,完全恢復單一CMC催化劑的活性。這充分表明,SiO2的存在抑制了CMC氧化物活性相的形成,導致催化劑失活。
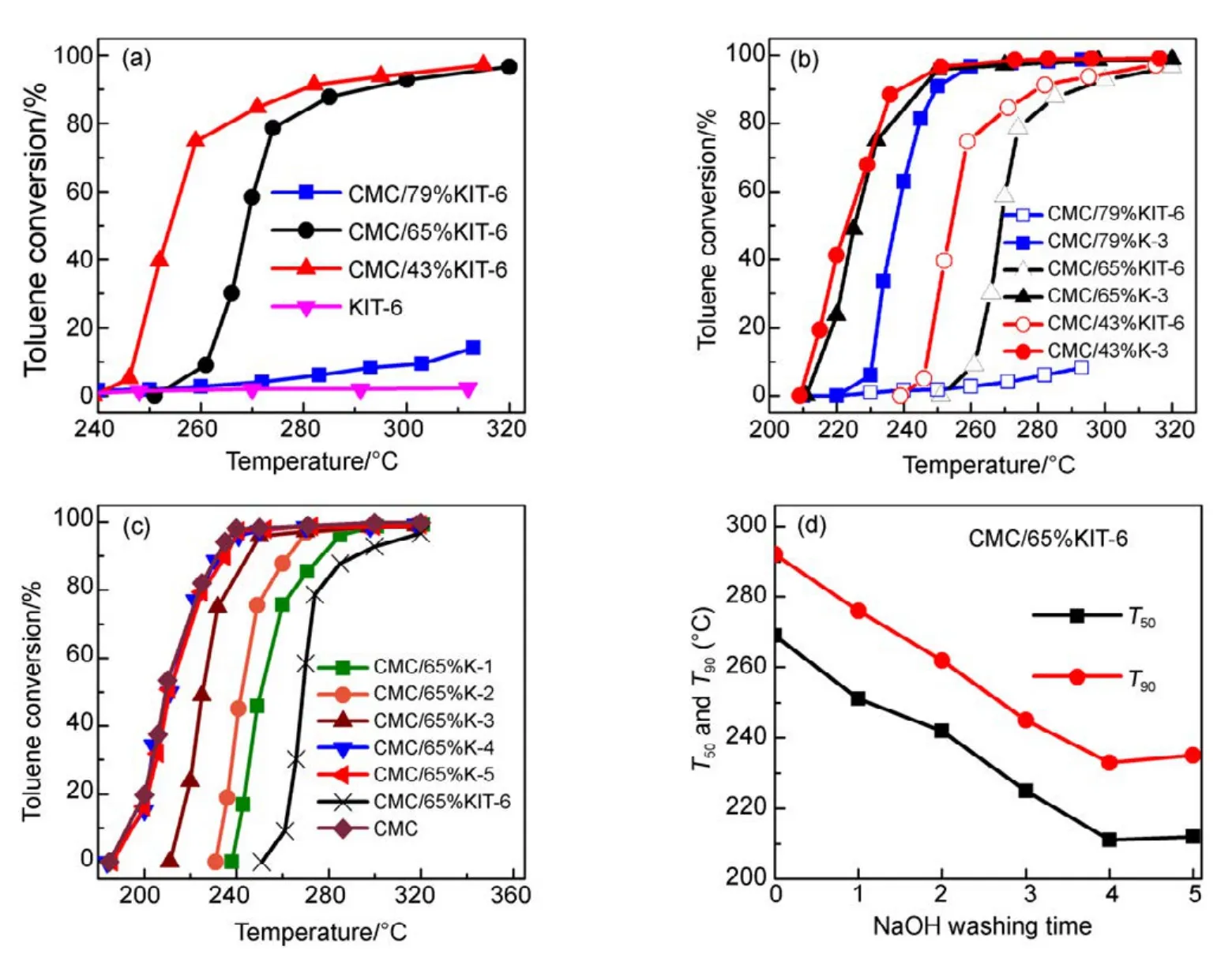
圖1 KIT-6載體對CMC復合氧化物催化活性影響Fig.1 Effect of KIT-6 on catalytic activity of CMC catalysts.
同樣,把CMC催化劑負載在同為SiO2基材的SBA-15和硅膠(silica)表面,測試了不同 CMC負載量催化劑的催化燃燒甲苯的特性(見圖2),發現隨著SiO2載體量增加,同樣出現迅速失活的現象(圖2(a, c))。采用70 °C NaOH堿洗三次,把SiO2去除掉,無論是硅膠還是SBA-15,其催化活性均得到一定程度的恢復。這說明只要是SiO2為基底的載體,均會導致鈰基復合氧化物催化劑失活。
3.2 SiO2對復合氧化物物相織構影響
為進一步研究SiO2載體對CMC結構的影響,圖3和表1給出了65%載體量的催化劑在不同堿洗次數后樣品的XRD表征數據。發現經過1、3、5次減洗后,CMC催化劑主晶相仍為立方相的CeO2,催化劑的晶胞參數與純的CeO2(0.5410 nm)相比均減小,晶粒大小和峰的強度均沒有變化,文獻28報道這是由于 Cu、Mn離子進入了 CeO2晶格中發生了同晶取代的結果,因為 Cu、Mn離子的半徑(Cu2+:0.072 nm、Cu+:0.077 nm;Mn4+:0.056 nm、Mn3+:0.062 nm、Mn2+:0.080 nm)均小于Ce離子的半徑(Ce4+:0.092 nm)。這說明堿洗后的催化劑仍然為銅錳鈰的固溶體結構28,SiO2并沒有改變 CMC復合氧化物的物相結構,CMC本征結構與載體SiO2量沒有關系。
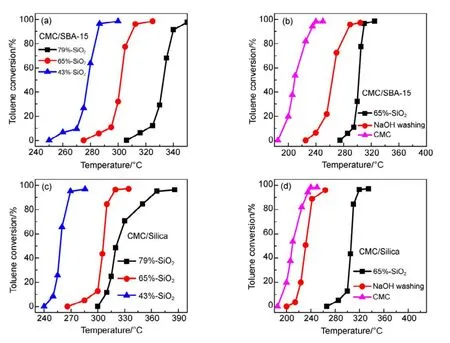
圖2 SBA-15和硅膠載體對CMC復合氧化物催化活性影響Fig.2 Effect of SBA-15 and Silica on the catalytic activity of CMC catalysts.
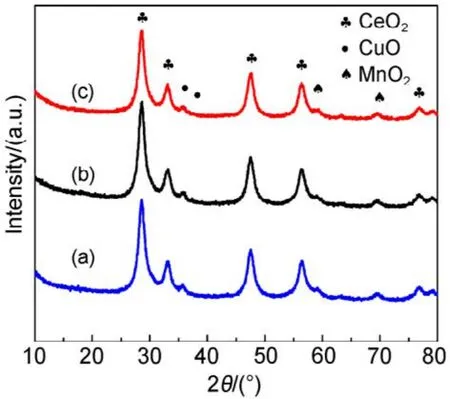
圖3 不同堿洗次數的CMC/65%KIT-6催化劑XRD圖譜Fig.3 XRD patterns of CMC/65%KIT-6 catalysts after NaOH washing.
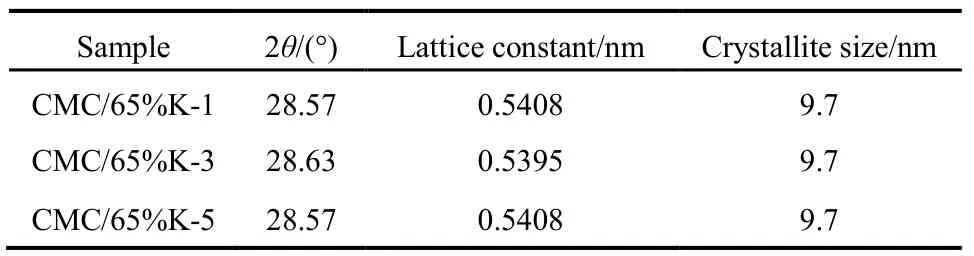
表1 不同堿洗次數的CMC/65%KIT-6催化劑的晶體結構Table 1 Crystal structure of CMC/65%KIT-6 catalysts.
圖4(a)和圖4(b)分別為KIT-6載體和不同堿洗次數的CMC/65%KIT-6催化劑的吸脫附等溫線和孔徑分布圖。表 2為載體和不同堿洗次數的CMC/65%KIT-6催化劑的比表面積和孔容數據,通過堿洗后催化劑比表面積大幅度減小,表明CMC/65%KIT-6催化劑通過簡單的堿洗基本除去了 KIT-6載體。但隨著堿洗次數的增加,SiO2的含量繼續減少,CMC/65%K-N催化劑的等溫線基本沒有變化,都屬于第IV類型吸附等溫線,具有H3型滯后環,介孔結構以及部分堆積孔結構,且催化劑的比表面積和孔容無明顯變化,這表明微量SiO2對CMC復合氧化物的比表面積、孔容等織構性質沒有大影響。
圖5為不同堿洗次數的CMC/65%KIT-6催化劑的HRTEM電鏡掃描圖,由圖可知,去除SiO2后的催化劑仍然具有CMC復合氧化物晶相結構,催化劑中主要成分仍然以CeO2晶相(d = 0.312 nm)和少量的CuOx(0.253 nm)和MnOx(0.187 nm)29組成。結合XRD表征,說明了Cu、Mn離子并沒有全部進入CeO2晶格中,部分 Cu-Mn在催化劑表面形成了混合晶相結構。通過表面能譜分析,隨著堿洗次數從1次增加到5次,催化劑中SiO2含量的均值由3.41%下降到1.22%,結果如圖5(a?c)所示。由圖5(d?f)可知,隨著堿洗次數增加,SiO2含量減少,CMC復合氧化物相變為明顯,堿洗5次后基本呈現復合氧化物混合晶相,這表明,SiO2不會改變CMC復合氧化物的晶相結構,通過堿洗去除SiO2后,CMC復合氧化物的活性位就會重新暴露出來,催化劑的活性得到恢復,這與甲苯催化燃燒的活性規律也是相一致的。
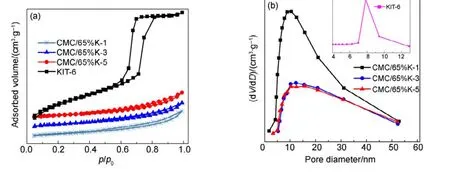
圖4 不同堿洗次數的CMC/65%KIT-6催化劑及載體的N2吸脫附等溫線和孔徑分布圖Fig.4 N2 adsorption-desorption isotherm and pore size distribution for CMC/65%KIT-6 catalysts after NaOH washing and KIT-6.

表2 不同堿洗次數CMC/65%KIT-6催化劑比表面積與孔結構Table 2 Surface area and pore characterization of CMC/65%KIT-6 catalysts after NaOH washing.

圖5 不同堿洗次數的CMC/65%KIT-6催化劑的HRTEM圖Fig.5 HRTEM images of CMC/65%KIT-6 catalysts with different treating times.
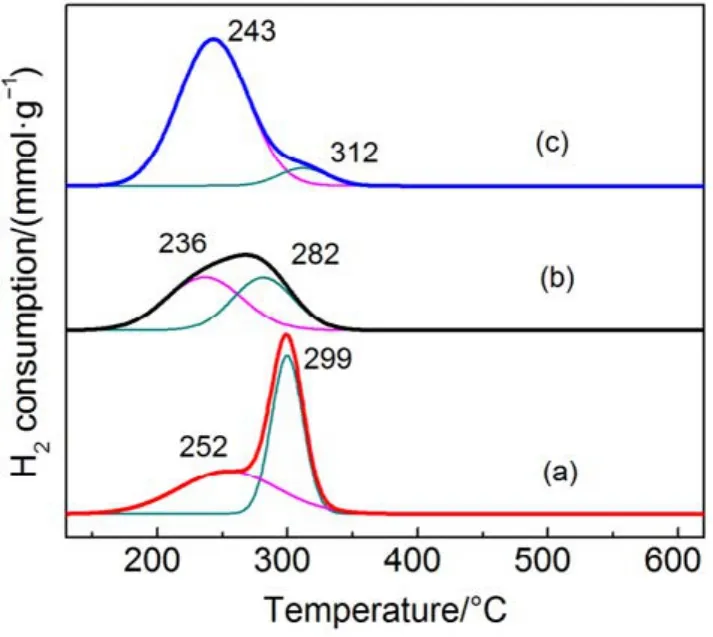
圖6 不同催化劑的H2-TPR圖譜Fig.6 H2-TPR profiles of different catalysts.
3.3 SiO2對催化劑氧化-還原性能的影響
圖6為不同載體量催化劑的H2-TPR譜圖,表 3為相應催化劑的還原峰的溫度以及耗氫量,圖中催化劑在測試時保持活性物質CMC量不變。由圖6可知,當載體量從43%增加到79%,低溫還原峰從252 °C偏移至243 °C,且低溫還原峰面積逐漸增加,低溫耗氫量從1.60 mmol·g?1增加至3.27 mmol·g?1,但催化劑的活性降低,根據文獻30報道,催化劑的H2-TPR還原溫度越低,其氧化還原性能越佳。但在這里卻發現完全相反的結果,CMC/79%-KIT-6雖然具有更低的還原溫度,并且其活性氧數量大,但催化活性卻表現不佳,甚至出現明顯的失活(見圖 1(a))。而 CMC/43%-KIT-6高溫還原峰面積大,低溫還原峰面積小,卻表現出高的活性。對于復合氧化物催化劑參與氧化還原反應,其活性氧起到整個氧化過程中關鍵性作用。一般我們把活性氧種類分成兩個種類,一是表面活性氧,二是體相的晶格氧。從圖 6分類,低溫還原峰應為表面氧脫除,高溫還原峰為體相的晶格氧脫除。因此,在SiO2載體含量大的催化劑中,復合氧化物高度分散,表面氧數量更多,而SiO2載體含量小的催化劑中,復合氧化物存在聚集,顯示出更多的晶格氧數量。而這些晶格氧對整個催化完全氧化過程起到更為關鍵的作用。
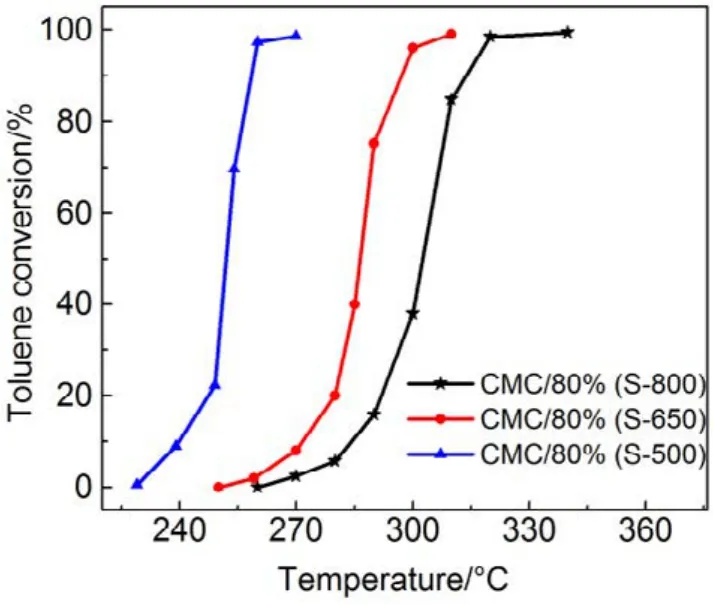
圖7 CMC/80%(S-T)催化劑催化燃燒甲苯活性曲線Fig.7 Activity curves of toluene combustion over CMC/80%(S-T) catalysts.
3.4 SiO2載體效應機制驗證
為了驗證SiO2對催化劑的載體效應,我們將硅膠(SiO2)載體在500、650、800 °C下分別焙燒3 h (標記為 Silica-T,T為溫度),以此來調節 SiO2比表面積大小和表面羥基數量,并把焙燒后的SiO2作為 CMC催化劑載體(標記為 CMC/80%(S-T),S為硅膠,T為溫度),考察其催化燃燒甲苯特性,催化燃燒結果如圖7所示,800 °C焙燒的硅膠負載催化劑活性要明顯好于500 °C焙燒載體的催化劑活性。對樣品進行BET分析(表4),發現焙燒溫度由500 °C升高到800 °C,載體的比表面積和孔容有所下降,但變化不大。對樣品進行H2-TPR表征(圖8),發現800 °C焙燒的硅膠載體高溫還原峰的峰面積更大。同時,對CMC/80%(S-500)和 CMC/80%(S-800)兩個樣品進行XPS表征(圖9),O 1s結果如表5所示,從中可知,CMC/(80%S-800)催化劑中晶格的含量為0.22,CMC/80%(S-500)催化劑中晶格氧的含量為0.12,結果表明CMC/80%(S-800)催化劑中晶格氧物種含量更多。通過對硅膠進行TG表征(圖10),以及硅膠高溫焙燒前后的紅外譜圖(圖 11),可以看出硅膠經過高溫焙燒后,其表面的羥基大量減少。綜合上述表征結果,可以發現硅膠載體經過高溫焙燒后,消除載體表面的羥基,使CMC的分散度下降,提高催化劑體相中晶格氧的含量,與表面氧相比,晶格氧需在更高的溫度被還原,因此其H2-TPR還原峰往高溫移動。這充分說明羥基數量的減少,更有利于復合氧化物表面構建具有豐富晶格氧的活性相。由此說明,在復合氧化物催化甲苯燃燒中,復合氧化物中的晶格氧是至關重要的,SiO2表面豐富的羥基不利于催化劑的催化活性,會導致CMC復合氧化物高度分散在SiO2表面,使活性氧物種主要以表面氧物種形式存在,降低CMC復合氧化物的催化活性。
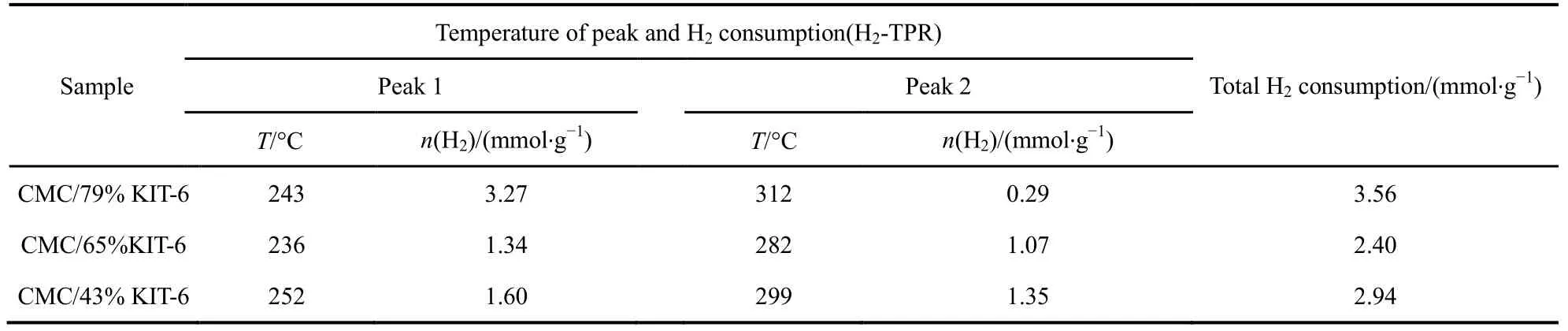
表3 不同載體量的催化劑的H2-TPRTable 3 H2-TPR of catalysts of different carrier weight.

表4 不同焙燒溫度的硅膠及CMC/80%SiO2催化劑的比表面積和孔道表征Table 4 Specific surface area and pore characterization of silica and CMC/80%SiO2 catalysts.

表5 不同催化劑的的O 1s結合能Table 5 O 1s binding energies of different catalysts.
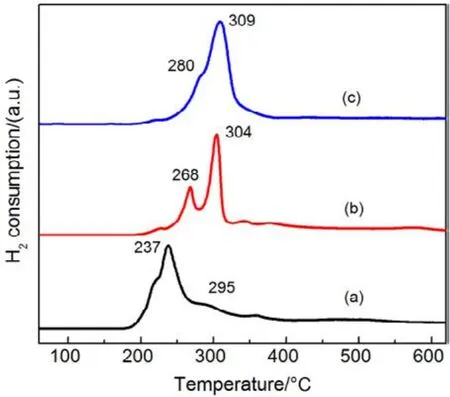
圖8 CMC/ 80%( S-T)催化劑的H2-TPR圖譜Fig.8 H2-TPR profiles of CMC/ 80%( S-T) catalysts.
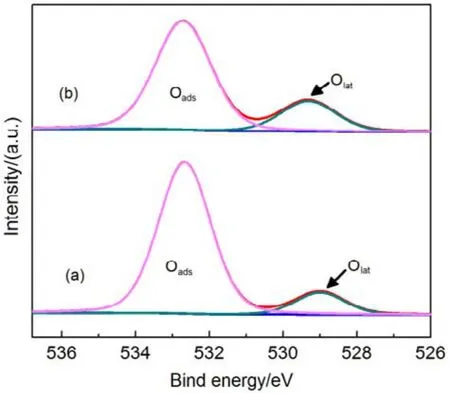
圖9 CMC/ 80%( S-T)催化劑的XPS圖譜Fig.9 XPS spetra of CMC/80%(S-T) catalysts.
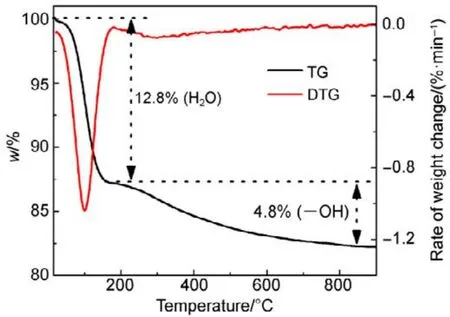
圖10 硅膠的TG和DTG曲線圖Fig.10 TG and DTG curves of silica.
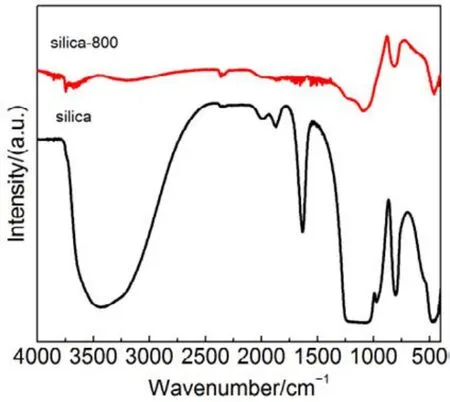
圖11 高溫焙燒后的硅膠的傅里葉變換紅外光譜圖Fig.11 FTIR spectra of silica gel after high temperature roasting.
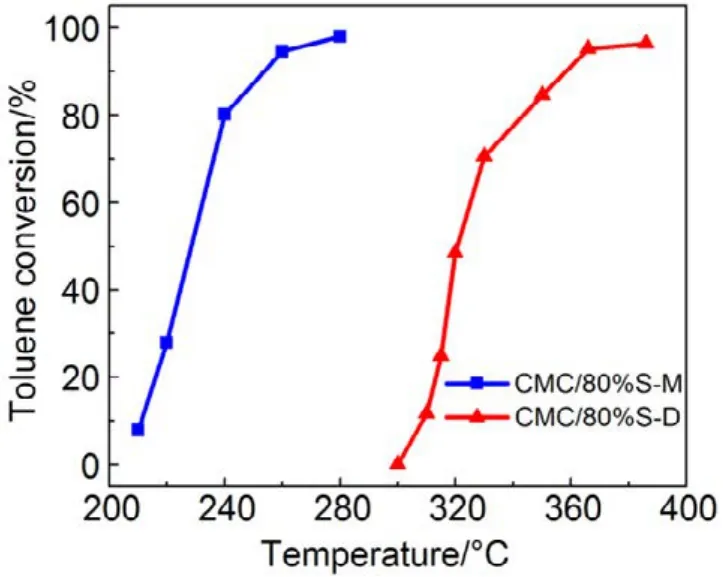
圖12 機械混合和浸漬法制備催化劑催化燃燒甲苯Fig.12 Toluene combustion over catalysts prepared with different methods.
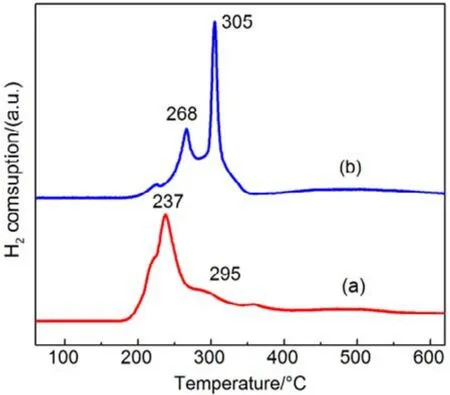
圖13 機械混合和浸漬法制備催化劑H2-TPR圖Fig.13 H2-TPR profiles of catalysts prepared with different methods.
同樣,為進一步驗證 SiO2表面羥基和高分散影響,我們把CMC復合氧化物與硅膠(SiO2)機械混合得到的催化劑,標記為 CMC/80%S-M,并將其與浸漬法制備的催化劑(標記為CMC/80%S-D)進行活性對比,結果如圖12所示,采用機械混合法制備的負載型催化劑的活性有了很大的提升,甲苯完全燃燒由365 °C降低至260 °C,而其低溫還原峰面積更小,高溫還原峰面積更高,體相中的晶格氧含量更多,如圖13所示,這與上文的結果都是相一致的。這進一步證明了 SiO2表面的羥基對復合氧化物活性的抑制作用,復合氧化物的高度分散反而會影響晶格氧生成,降低復合氧化物的催化活性。
4 結 論
鈰基復合氧化物與載體或模板劑SiO2表面接觸,并不會改變鈰基復合氧化物催化劑的物理結構,但SiO2表面豐富的羥基會使低負載量的鈰基氧化物在表面高度分散,導致復合氧化物晶格氧數量減少,表面氧數量增加,催化活性迅速下降。通過堿洗去除SiO2或高溫焙燒去除SiO2表面羥基,都可以使負載的鈰基復合氧化物保持豐富的晶格氧數量,恢復高的催化氧化活性。
(1) Mesa, M.; Sierra, L.; Patarin, J.; Guth, J. L. Solid State Sci. 2005, 7(8), 990. doi: 10.1016/j.solidstatesciences.2005.04.006
(2) Tsung, C. K.; Fan, J.; Zheng, N.; Shi, Q.; Forman, A. J.; Wang, J.;Stucky, G. D. Angew. Chem. Int. Ed. 2008, 47 (45), 8682.doi: 10.1002/anie.200802487
(3) Brezesinski, T.; Groenewolt, M.; Antonietti, M.; Smarsly, B. Angew.Chem. Int. Ed. 2006, 45 (5), 781. doi: 10.1002/anie.200502332
(4) Kresge, C. T.; Leonowicz, M. E.; Roth, W. J.; Vartuli, J. C.; Beck, J.S. Nature 1992, 359 (6397), 710. doi: 10.1038/359710a0
(5) Hammami, R.; Aissa, S. B.; Batis, H. Appl. Catal. A-Gen. 2009, 353(2), 145. doi: 10.1016/j.apcata.2010.05.048
(6) Beck, J. S.; Vartuli, J. C.; Schmitt, K. D.; Olson, D.H.; Sheppard, E.W.; McCullen, S. B.; Higgins, J. B.; Schlenker, J. L.; Roth, W. J.;Leonowica, M. E.; Kresge, C. T; Chu, C. T. W. J. Am. Chem. Soc.1992, 114 (27), 10834. doi: 10.1021/ja00053a020
(7) Ying, J. Y.; Mehnert, C. P.; Wong, M. S. Angew. Chem. Int. Ed.1999, 38 (1?2), 57. doi: 10.1002/(SICI)1521-3773(19990115)38:1/2<56::AID-ANIE56>3.0.CO;2-E
(8) Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G. H.;Chmelka, B. F.; Stucky, G. D. Science 1998, 279, 548. doi: 10.1126/science.279.5350.548
(9) Wang, Y. G.; Wang, Y. Q.; Liu, X. H.; Guo, Y.; Guo, Y. L.; Lu, G. Z.J. Nanosci. Nanotechno. 2009, 9 (2). 933.doi: 10.1166/jnn.2009.C057
(10) Asefa, T.; Lennox, R. B. Chem. Mater. 2005, 17 (10), 2481.doi: 10.1021/ cm047800j
(11) Kleitz, F.; Choi, S. H.; Ryoo, R. Chem. Commun. 2003, 9 (17), 2136.doi: 10.1039/b306504a
(12) Kleitz, F.; Liu, D.; Anilkumar, G. M.; Park, I. S.; Solovyov, L. A.;Shmakov, A. N.; Ryoo, R. J. Phys. Chem. B 2003, 107 (51), 14296.doi: 10.1021/jp036136b
(13) Ryoo, R.; Kim, J. M.; Ko, C. H.; Shin, C. H. J. Phys. Chem. 1996,100 (45), 17718. doi: 10.1021/jp9620835
(14) Dickinson, C.; Zhou, W.; Hodgkins, R. P.; Shi, Y.; Zhao, D.; He, H.Chem. Mater. 2006, 18 (13), 3088. doi: 10.1021/cm060014p
(15) Yue, X. L.; Liu, L.; Zhang, M.; Yang, M.; Dong, Y. L.; Cheng, M.J. Chinese J. Catal. 2009, 30 (2), 95. [岳響玲, 柳 林, 張 敏,董永來, 程謨杰. 催化學報, 2009, 30 (2), 95.]doi: 10.3321/j.issn:0253-9837.2009.02.005
(16) Zhu, J. J.; Xiao, D. H.; Li, J.; Yang, X. G. Catal. Lett. 2009, 129(1?2). 240. doi: 10.1007/s10562-008-9807-8
(17) Laha, S. C.; Ryoo, R. Chem. Commun. 2003, 9 (17), 2138.doi: 10.1039/b305524h
(18) Yang, H.; Zhao, D. J. Mater. Chem. 2005, 15 (12), 1217.doi: 10.1039/b414402c
(19) Wang, Y.; Yang, C. M.; Schmidt, W.; Spliethoff, B.; Bill, E.; Schüth,F. Adv. Mater. 2005, 17 (1), 53. doi: 10.1002/adma.200400777
(20) Jun, S.; Sang Hoon, J.; Ryoo, R.; Kruk, M.; Jaroniec, M.; Liu, Z.;Ohsuna, T.; Terasaki, O. J. Am. Chem. Soc. 2014, 122 (43), 10712.doi: 10.1021/ja002261e
(21) Fan, J. X.; Yang, S.; Qian, W.; Shi, F. T.; Huang, H. F. Chin. Med.J.-Peking 2016, 129 (7). 785.doi: 10.4103/0366-6999.178954
(22) Jermy, B. R.; Kim, S. Y.; Bineesh, K. V.; Selvaraj, M.; Park, D. W.Korean J. Chem. Eng. 2009, 26 (5), 1235.doi: 10.1007/s11814-009-0199-2
(23) Tüysüz, H.; Salaba?, E. L.; Weidenthaler, C.; Schüth, F. J. Am. Chem.Soc. 2008, 130 (1), 280. doi: 10.1021/ja075528j
(24) Mizoshita, N.; Tani, T.; Inagaki, S. Chem. Soc. Rev. 2011, 40 (2),789. doi: 10.1039/c0cs00010h
(25) Zhang, J.; Zhang, G.; Gao, Y.; Sun, R.; Wong, C. P. J. Mater. Sci.2016, 51 (17). 7966. doi: 10.1007/s10853-016-0066-6
(26) Kim, T. W.; Kleitz, F.; Paul, B.; Ryoo, R. J. Am. Chem. Soc. 2005,127 (20), 7601. doi: 10.1021/ja042601m
(27) Kleitz, F.; Bérubé, F.; Guillet-Nicolas, R.; Yang, C. M.; Thommes,M. J. Phys. Chem. C 2010, 114 (20), 9344. doi: 10.1021/jp909836v
(28) Lu, H.; Huang, J.; Zhou, Y.; Zhu, Q.; Chen, Y. CIESC Journal 2015,66 (6), 2105. [盧晗鋒, 黃金星, 周 瑛, 朱秋蓮, 陳銀飛. 化工學報, 2015, 66 (6), 2105.] doi: 10.11949/j.issn.0438-1157.20141930
(29) Okumura, K.; Kobayashi, T.; Tanaka, H.; Niwa, M. Appl. Catal.B-Environ. 2003, 44 (4), 325. doi: 10.1016/s0926-3373(03)00101-2
(30) Tang, W.; Wu, X.; Li, S.; Shan, X.; Liu, G.; Chen, Y. Appl. Catal.B-Environ. 2015, 162, 110. doi: 10.1016/j. apcatb.2014.06.030
Deactivation Mechanism of CeO2-Based Mixed Oxide Catalysts Supported on SiO2
ZHAN Lin-Jun SUN Xiao-Yan ZHOU Ying*ZHU Qiu-Lian CHEN Yin-Fei LU Han-Feng
(College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China)
Here we reported the effect of the Cu-Mn-Ce-SiO2(CMC-SiO2) interaction on the physical and chemical aspects of the catalytic combustion of toluene by adjusting the loading amount of the CMC mixed oxide on SiO2. Notably, the CMC/KIT-6 catalyst with low CMC loading performed poorly with an obvious deactivation, owing to the inhibition of the metal oxides active sites, while the activity recovered after washing away some SiO2. The catalysts were characterized by X-ray diffraction (XRD), H2temperature-programmed reduction (H2-TPR), N2adsorption, and high-resolution transmission electron microscopy (HRTEM). Although there is no change in crystal structure after loading on SiO2, active oxygen species immigrate from lattice to surface for SiO2surface rich in hydroxyl groups and having high dispersion of CMC, leading to deactivation of the CMC catalyst. However, it is worth mentioning that the lattice oxygen played a key role in catalytic combustion. The activity of the CMC catalyst recovered when the quantity of lattice oxygen increased upon removing surface ―OH groups by calcination or removing some SiO2by alkali washing.
SiO2; Cu-Mn-Ce mixed oxide; Deactivation; Supporting effect; Catalytic combustion
January 13, 2017; Revised: March 10, 2017; Published online: March 31, 2017.
O643
10.3866/PKU.WHXB201703312 www.whxb.pku.edu.cn
*Corresponding author. Email: wifx@zjut.edu.cn; Tel: +86-571-88320767.
The project was supported by the National Natural Science Foundation of China (21506194, 21676255), and Natural Science Foundation of Zhejiang Province, China (Y14E080008, Y16B070025).
國家自然科學基金(21506194, 21676255)和浙江省自然科學基金(Y14E080008, Y16B070025)資助項目
? Editorial office of Acta Physico-Chimica Sinica
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
