生物芯片高通量檢測卵巢癌轉錄組差異表達基因的生物標記識別
2017-11-22 01:45:06何文龍隋杏玲
臨床檢驗雜志(電子版) 2017年1期
何文龍 隋杏玲
生物芯片高通量檢測卵巢癌轉錄組差異表達基因的生物標記識別
何文龍 隋杏玲
目的探究卵巢癌中基因的表達情況并識別重要的代謝通路。方法從ArrayExpress數據庫下載卵巢癌芯片數據,使用rank product方法識別差異表達基因;利用STRING900、STRING、Bossi&Lehner和PSICQUIC四個數據庫對這些差異基因進行KEGG通路富集分析,并運用R語言運算取這些通路的交集,以找出在卵巢癌發生中重要的代謝通路;最后構建各通路中重疊基因的網絡節點圖,以識別通路中的重要基因。結果在卵巢癌中共檢測到1,732個差異表達基因和176條代謝通路。其中,p53信號通路、酪氨酸代謝、鞘脂類代謝是卵巢癌中最重要的代謝通路。這些通路中基因的表達均發生了顯著變化。結論所識別的重要代謝通路和這些通路中基因表達的變化可作為卵巢癌診斷的潛在生物標記。
卵巢癌;差異表達基因;通路富集分析;網絡構建
作者單位:300010 天津,天津醫科大學公共衛生學院(何文龍);250101 濟南,濟南基因云館數字圖書館(隋杏玲)(通訊作者:隋杏玲)
卵巢癌惡性腫瘤是危害女性健康與生命的惡性腫瘤之一,僅有20%左右的卵巢癌患者可在早期被發現,晚期患者的死亡率居婦科惡性腫瘤死亡率之首[1,2]。
目前,卵巢癌的早期診斷多采用血清CA125、癌胚抗原(carcino-embryonic antigen,CEA)、人附睪蛋白4(human epididymis protein-4,HE4)等作為重要標記物。但這些標記物普遍存在敏感性和特異性差、準確率較低等問題[3],遠不能滿足臨床實際的應用需要。代謝組學作為一種將圖像識別方法和生物信息學結合起來的分析技術,在卵巢癌的早期診斷、預后和治療評價方面得到了廣泛的應用并已檢測出了可作為卵巢癌診斷的候選標記物[4,5]。雖然這些研究為卵巢癌的生物學特性的異常改變及早期診斷提供了途徑,但對于卵巢癌發病的分子機理及各基因間的相互作用還不能清晰闡釋。已有的研究表明,卵巢癌的發生和發展是一個復雜的過程,具有復雜的生物學行為,涉及了多個基因的異常表達和相互作用。因此,尋找更加靈敏、特異、檢測方便的卵巢癌腫瘤標記物,對于早期發現并提高治療效果具有重要意義。
高通量技術和生物信息學分析手段的聯合應用,為從基因組水平研究疾病的分子基礎提供了途徑。本文運用生物信息學方法,對從ArrayExpress數據庫選取的卵巢癌的微陣列芯片基因表達數據進行挖掘,旨在通過對差異表達基因進行通路富集分析,找出卵巢癌中重要的代謝通路,并對其中包含的基因進行分析,以期為卵巢癌的診斷提供分子標記。
1 材料與方法
1.1 數據收集與預處理 實驗所用數據芯片E-GEOD-10971[6]、E-GEOD-14001[7]、E-GEOD-18520[8]和E-GEOD-27651均選自ArrayExpress數據庫(http://www.ebi.ac.uk/arrayexpress/)。使用Affymetrix包讀取每個芯片數據,分別采用穩健多陣列平均法(RMA算法)[9]和分位數法[10]對其進行背景矯正與規范化,以消除非特異性雜交的影響;運用mas5算法[11]對探針的匹配值與誤配值進行修正,對于表達式的值則采用中位數方法進行取值。使用GeneFilter包的featureFilter方法進行數據篩選,去掉四分位數間距大于0.5的基因,若有多個探針對應同一個基因,則保留其中間值。
1.2 差異表達基因的篩選 使用RankProd包中的rank product方法對4個芯片數據進行合并,然后計算各基因的FoldChange(FC)值。|log2FC|>2且P小于0.01的基因則視為差異表達基因,并區分出上調與下調的基因。
1.3 通路富集分析 利用在線分析軟件EnrichNet-Network-based enrichment analysis(http://www.enrichnet.org/)中的STRING900、STRING、Bossi&Lehner和PSICQUIC分子網絡數據庫對所得的|log2FC|≥3的差異表達基因進行通路富集分析(Kyoto Encyclopedia of Genes and Genomes,KEGG),然后運用R語言運算取所有通路的交集,并計算每條通路的Xd值[12]的平均值,以找出在卵巢癌發生中比較重要的通路。
1.4 通路網絡構建 從http://www.enrichnet.org/下載Xd值的平均值大于0.68的各通路的cytoscape-network數據,將其導入Cytoscape 3.1.0軟件中構建它們的網絡節點圖,僅保留該通路基因集中的重疊基因,然后將四個數據庫中同一通路的網絡節點圖進行合并,找出該通路的重要基因。
2 結果
2.1 差異表達基因的識別 通過對所得基因芯片數據進行過濾篩選,共得到1,732個差異表達基因,其中上調基因580個,下調基因1,152個。前27個上調和前29個下調的差異表達基因的具體信息見表1。
2.2 通路富集分析 將|log2FC|≥3的656個差異表達基因輸入STRING900、STRING、Bossi&Lehner和PSICQUIC四個數據庫進行在線KEGG生物通路注釋,分別得到了193條、196條、177條和192條通路。進一步通過R語言運算取交集后,共得到176條通路。比較重要的通路有p53信號通路、酪氨酸代謝、鞘脂類代謝、甘氨酸-絲氨酸-蘇氨酸代謝、細胞外基質相互作用等。前10條通路的具體信息見表2。
2.3 基因的相互作用網絡分析 Cytoscape作為一個強大的分子相互作用網絡分析和可視化軟件,對生物信息學的研究具有重要幫助。連通度作為評價節點中心性的參數,其大小可以反映該節點在網絡中的重要性。通過Cytoscape軟件對卵巢癌中重要通路的差異表達基因進行網絡圖譜分析可以發現,細胞周期蛋白依賴性激酶抑制劑2A(CDKN2A)在p53信號通路中與細胞周期蛋白(CCND2、CCNE1)和蛋白磷酸酶(PPM1D)密切相關,暗示該基因在p53信號通路改變中具有重要作用(圖1A);單胺氧化酶(MAOA、MAOB)和乙醇脫氫酶(ADH1B)在酪氨酸代謝通路中起著中心性作用(圖1B);在鞘脂類代謝通路中主要包含5個基因,各基因間緊密作用,共同調控疾病的發生(圖1C)。各通路中基因間的相互聯系及其具體參數見圖1和表3。
3 討論
作為女性腫瘤的高發疾病,卵巢癌的發生是多種體內多種因素共同作用的結果。我們通過對卵巢癌基因表達芯片進行分析,發現共有1,732個基因的表達發生了變化;進一步利用不同數據庫對部分差異表達基因進行通路富集分析表明,p53信號通路、酪氨酸代謝通路和鞘脂類代謝通路是卵巢癌發生中的重要通路,暗示這些通路的改變可能是卵巢癌發生、發展的重要原因之一。這與Fu等[13]的研究結果基本一致。
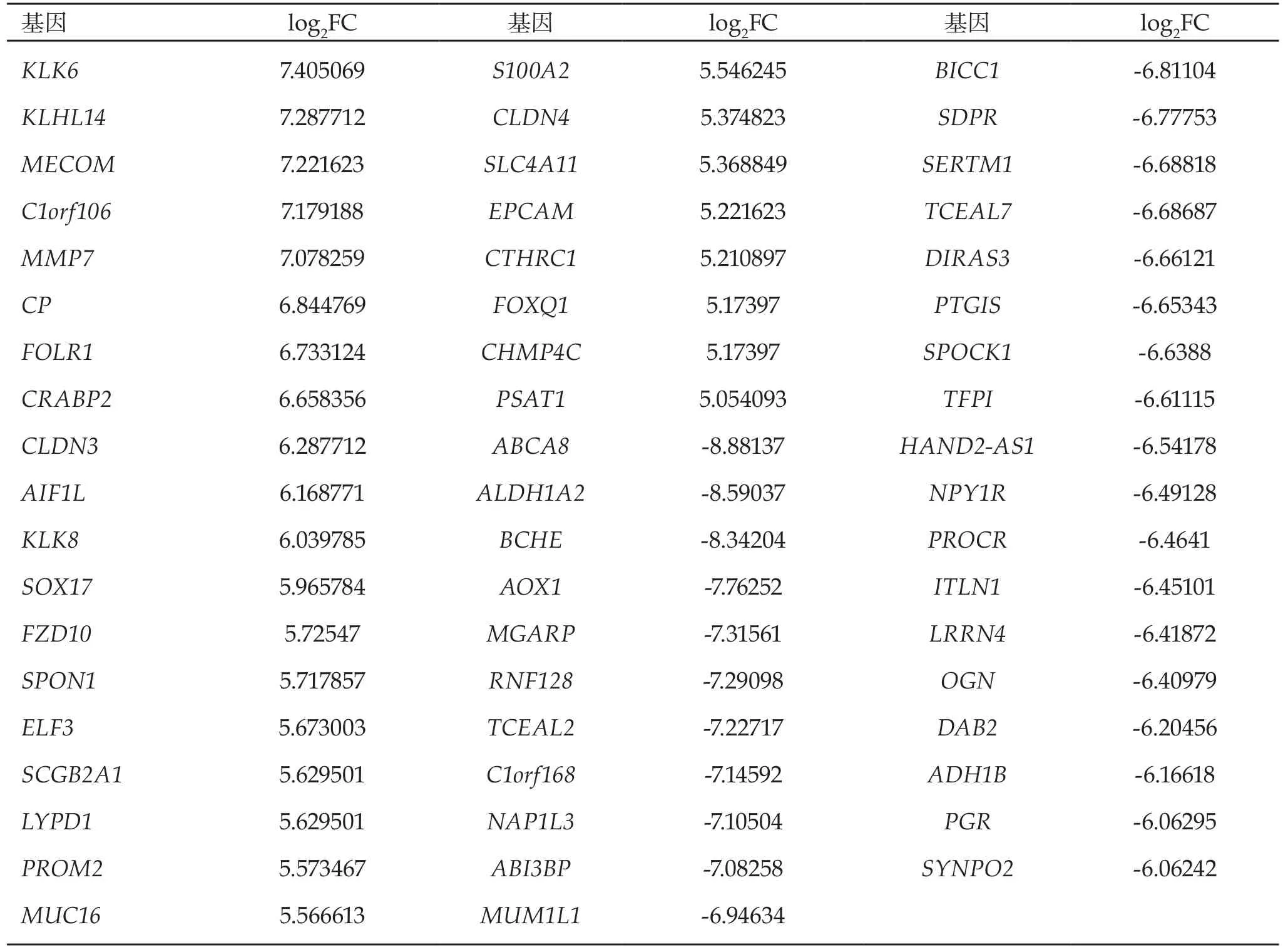
表1 卵巢癌中P值小于0.01的前27個上調和前29個下調的差異表達基因
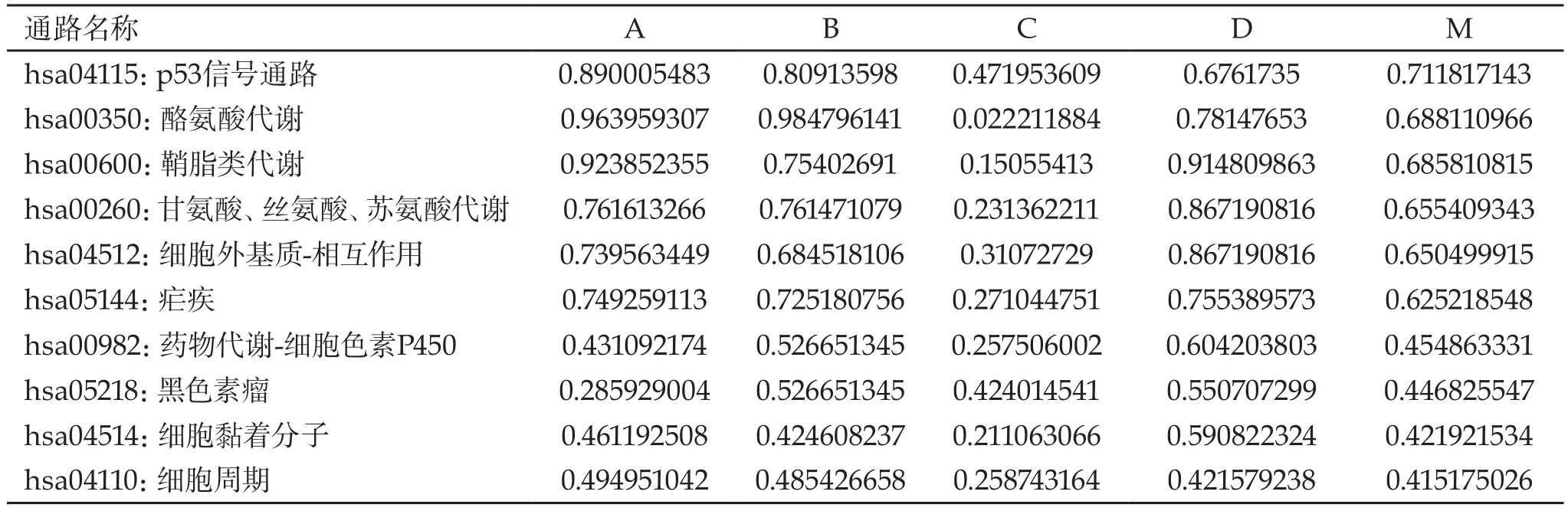
表2 基于KEGG的通路富集分析
已知p53通路的異常改變是腫瘤細胞惡變轉化的主要原因之一。本研究通過對參與p53信號通路的基因的網絡分析發現,CDKN2A在p53信號通路起著重要作用。作為與細胞周期相關的抑癌基因,CDKN2A/p16在人類癌癥的發生中經常發生突變[14],但在早期卵巢癌和轉移性卵巢癌中發生突變的頻率卻很低[15]。Bian等[16]利用DNA重組技術構建了人類卵巢癌細胞系UACC-1598的CDKN2A/p16-A148T突變體和CDKN2A/p16-野生型基因表達體系,發現在卵巢癌中CDKN2A/p16的第148位密碼子發生了單堿基突變。Kim等[17]在早期卵巢癌和復發性上皮性卵巢癌也發現了該基因的突變。在本研究中,CDKN2A(pfp=3.671164,P=0)是差異表達基因之一,其表達量上調可能是導致p53通路改變的重要原因。

表3 各通路中差異表達基因的信息
酪氨酸作為苯丙氨酸代謝的產物之一,其代謝的異常可引起疾病的發生。Fong等[4]通過對卵巢癌患者的卵巢代謝物進行分析發現,苯丙酮酸和苯乳酸的含量明顯升高;卵巢癌患者在接受治療后,血清中酪氨酸-賴氨酸-亮氨酸-40的含量明顯下降[18]。卵巢癌中酪氨酸代謝的異常可能與苯丙氨酸代謝密切相關。我們的研究結果表明,酪氨酸代謝是卵巢癌中重要的代謝通路之一,與酪氨酸下游產物代謝相關的基因發生了明顯下調。研究表明,卵巢癌治療藥物藥效的發揮與酪氨酸激酶受體的下調有關[19]。目前已有利用酪氨酸激酶定向治療卵巢癌的報道,盡管它的療效還無定論[20]。這些結果暗示,酪氨酸代謝可作為卵巢癌診斷的指標之一。
鞘脂類作為細胞膜的組成成分之一,其代謝產物神經酰胺、神經鞘氨醇和1-磷酸鞘氨醇在腫瘤的發生發展中發揮著重要作用,它們可以調節細胞的增值、存活及凋亡等。Babahosseini等[21]的研究發現,神經酰胺和1-磷酸鞘氨醇能夠降低發生惡變的小鼠卵巢上皮細胞的彈性,而鞘氨醇的作用卻正好相反。Guillwrmet-Guibert等[22]發現,通過加入鞘氨醇激酶-1抑制劑、神經酰胺類似物等提高細胞內神經酰胺的水平或者降低細胞內鞘氨醇激酶-1的活性來提高神經酰胺、1-磷酸鞘氨醇的比率,可有效提高胰腺癌細胞對胞苷的化學敏感性,從而殺死癌細胞。Anderson等[23]研究表明,通過添加外源性鞘氨醇能夠使小鼠上皮性卵巢癌細胞中檸檬酸合成酶活性降低、促進三羧酸循環,減少膽固醇的合成和糖酵解,從而改變細胞代謝路徑。以上研究表明,鞘脂類代謝與腫瘤的發生、進展密切相關,它在腫瘤的治療中具有巨大的潛力。Choi等[24]利用小鼠動物模型研究發現α-半乳糖神經酰胺輔助腫瘤細胞疫苗治療卵巢癌,可打破腫瘤細胞的免疫耐性,明顯提高血清中γ-干擾素的水平。我們的通路富集分析結果表明,鞘脂類代謝在卵巢癌的發生發展中占有重要地位,與鞘脂類代謝相關的主要基因密切相互作用并都下調表達,這些基因的表達的異常可能是導致卵巢癌發生的重要原因。
綜上所述,本研究通過生物信息學方法對卵巢癌相關基因的表達和通路富集分析發現,在卵巢癌的發生發展中,有大量基因的表達發生了明顯變化(上調或下調),并引起了通路代謝的異常,p53信號通路、酪氨酸代謝通路和鞘脂類代謝通路可作為卵巢癌診斷的重要指標。
1 Teneriello MG,Park RC.Early detection of ovarian cancer.CA Cancer J Clin,1995,45(2): 71-87.
2 Jacobs IJ,Menon U.Progress and challenges in screening for early detection of ovarian cancer.Mol Cell Proteomics,2004,3(4): 355-366.
3 Williams TI,Toups KL,Saggese DA,et al.Epithelial ovarian cancer: Disease etiology,treatment,detection,and investigational gene,metabolite,and protein biomarkers.J Proteome Res,2007,6(8): 2936-2962.
4 Fong MY,McDunn J,Kakar SS.Identification of metabolites in the normal ovary and their transformation in primary and metastatic ovarian cancer.PLoS One,2011,6(5): e19963.
5 Fan L,Zhang W,Yin M,et al.Identification of metabolic biomarkers to diagnose epithelial ovarian cancer using a UPLC/QTOF/MS platform.Acta Oncol,2012,51(4):473-479.
6 Tone AA,Begley H,Sharma M,et al.Gene expression profiles of luteal phase fallopian tube epithelium fromBRCAmutation carriers resemble high-grade serous carcinoma.Clin Cancer Res,2008,14(13): 4067-4078.
7 Tung CS,Mok SC,Tsang YT,et al.PAX2 expression in low malignant potential ovarian tumors and low-grade ovarian serous carcinomas.Mod Pathol,2009,22(9):1243-1250.
8 Gamwell LF,Gambaro K,Merziotis M,et al.Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics.Orphanet J Rare Dis,2013,8: 33.
9 Ma L,Robinson LN,Towle HC.ChREBP.ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver.J Biol Chem,2006,281(39):28721-28730.
10 Rifai N,Ridker PM.Proposed cardiovascular risk assessment algorithm using high-sensitivity C-reactive protein and lipid screening.Clin Chem,2001,47(1): 28-30.
11 Zhang L,Miles MF,Aldape KD.A model of molecular interactions on short oligonucleotide miroarrays.Nat Biotechnol,2003,21(7): 818-821.
12 Glaab E,Baudot A,Krasnogor N,et al.EnrichNet:network-based gene set enrichment analysis.Bioinformatics,2012,28(18): i451-i457.
13 Fu LJ,Wang B.investigation of the hub genes and related mechanism in ovaian cancer via bioinformatics analysis.J Ovarian Res,2013,6(1): 92.
14 Witkiewicz AK,Knudsen KE,Dicker AP,et al.The meaning of p16 (ink4a) expression in tumors:functional significance,clinical associations and future developments.Cell Cycle,2011,10(15): 2497-2503.
15 Schuyer M,van Staveren IL,Klijn JG,et al.SporadicCDKN2(MTS1/p16ink4) gene alterations in human ovarian tumours.Br J Cancer,1996,74(7): 1069-1073.
16 Bian Z,Yu Y,Yang T,et al.Effect of tumor suppressor gene cyclin-dependent kinase inhibitor 2A wild-type and A148T mutant on the cell cycle of human ovarian cancer cells.Oncol Lett,2014,7(4): 1229-1232.
17 Kim YM,Lee SW,Chun SM,et al.Analysis and comparison of somatic mutations in paired primary and recurrent epithelial ovarian cancer samples.PLoS One,2014,9(6): e99451.
18 Choudhuri S,Sharma C,Banerjee A,et al.A repertoire of biomarkers helps in detection and assessment of therapeutic response in epithelial ovarian cancer.Mol Cell Biochem,2014,386(1-2): 259-269.
19 Cho YR,Choi SW,Seo DW.Thein vitroantitumor activity of Siegesbeckia glabrescens against ovarian cancer through suppression of receptor tyrosine kinase expression and the signaling pathways.Oncol Rep,2013,30(1): 221-226.
20 Morotti M,Becker CM,Menada MV,et al.Targeting tyrosine-kinases in ovarian cancer.Expert Opin Investig Drugs,2013,22(10): 1265-1279.
21 Babahosseini H,Roberts PC,Schmelz EM,et al.Bioactive sphingolipid metabolites modulate ovarian cancer cell structural mechanics.Integr Biol (Camb),2013,5(11):1385-1392.
22 Guillermet-Guibert J,Davenne L,Pchejetski D,et al.Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug.Mol Cancer Ther,2009,8(4): 809-820.
23 Anderson AS,Roberts PC,Frisard MI,et al.Metabolic changes during ovarian cancer progression as targets for sphingosine treatment.Exp Cell Res,2013,319(10):1431-1442.
24 Choi YS,Hoory T,Monie A,et al.Alpha-Galactosylceramide enhances the protective and therapeutic effects of tumor cell based vaccines for ovarian tumors.Vaccine,2008,26(46): 5855-5863.
Microarray data analysis and biomarker identification of ovarian cancer
Wenlong HE1,Xingling SUI2
1School of Public Health,Tianjin Medical University,Tianjin 300010,China;2Digital Library,Ji'nan Gene Cloud Museum,Ji’nan 250101,China
ObjectiveTo explore the gene expression pattern and identify the important metabolic pathways in ovarian cancer.MethodsThemicroarray data of ovarian cancer was downloaded from ArrayExpress database,and the differentially expressed (DE) genes in it were identified by the rank product method.The KEGG pathway enrichment analysis of these DE genes was performed by using the on line database STRING900,STRING,Bossi&Lehner and PSICQUIC.The R language was used to computing the intersection of these pathways and finding out the important pathways in ovarian cancer.Finally,the important genes and their relations in these pathways were analyzed by constructing the nodes network of the overlapped genes.ResultsA total of 1,732 DE genes and 176 metabolic pathways were involved in the occurrence.The p53 signaling pathway,tyrosine metabolism and sphingolipid metabolism were the most important pathways in ovarian cancer.The expression pattern of the genes that involved in these pathways was also greatly changed.ConclusionThe important pathways and change of the genes involved in them can be regard as the underlying biomarkers for the diagnosis of ovarian cancer.
Ovarian cancer; Differentially expressed genes; Pathway enrichment analysis; Network construction
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
財經(2017年2期)2017-03-10 14:35:35
山東工業技術(2016年15期)2016-12-01 05:31:22
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
