RP-HPLC同時測定射干9種活性成分含量
2017-12-27 06:30:35尤獻民鄒桂欣邸子真李國信
中國中醫藥信息雜志 2017年1期
尤獻民,鄒桂欣,邸子真,李國信
遼寧省中醫藥研究院,遼寧 沈陽 110034
RP-HPLC同時測定射干9種活性成分含量
尤獻民,鄒桂欣,邸子真,李國信
遼寧省中醫藥研究院,遼寧 沈陽 110034
目的 建立RP-HPLC同時測定射干藥材中芒果苷、射干苷、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、白射干素及次野鳶尾黃素共9種成分含量的方法。方法 采用LeapsilTMC18色譜柱(100 mm×2.1 mm,3 μm),流動相為乙腈-0.1%甲酸水溶液梯度洗脫,檢測波長265 nm,流速0.5 mL/min,柱溫40 ℃。結果 芒果苷、射干苷、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、白射干素、次野鳶尾黃素的線性范圍分別為0.214 0~2.568 μg(r=0.999 5)、0.437 0~5.244 μg(r=0.999 3)、0.460 0~5.520 μg(r=0.999 9)、0.078 40~0.940 8 μg(r=0.999 6)、0.138 0~1.656 μg(r=0.999 3)、0.051 00~0.612 0 μg(r=0.997 5)、0.113 0~1.356 μg(r=0.999 9)、0.051 63~0.619 6 μg(r=0.999 8)、0.151 0~1.812 μg(r=0.999 9),平均加樣回收率分別為97.73%、96.81%、97.78%、97.55%、96.86%、98.60%、97.77%、98.04%、97.89%,RSD分別為0.7%、1.1%、2.3%、2.1%、1.3%、1.4%、2.3%、1.6%、1.9%。應用該方法測定了5批射干藥材中9種成分的含量。結論 該方法準確、可靠,可為射干藥材的質量控制提供參考依據。
射干;活性成分;反相高效液相色譜法;含量測定
射干是鳶尾科射干屬植物射干Belamcanda chinensis(L.)DC.的干燥根莖,具有清熱解毒、利咽消痰、散血消腫的功效,是治療喉痹咽痛常用藥,被歷版《中華人民共和國藥典》收載。射干中主要含有異黃酮類化合物、醌類、酚類、二環三萜類化合物、甾類化合物及一些微量成分[1],其中黃酮類成分具有顯著的鎮痛、止咳、抗炎抑菌[2-3]和抗病毒[4]等多種藥理作用。據文獻報道,鳶尾苷和鳶尾黃素對大鼠腹腔巨噬細胞中前列腺素E2產物和環氧合酶-2誘導具有抑制作用[5],野鳶尾黃素對巨噬細胞內一氧化氮和前列腺素E2產物具有抑制作用[6],從射干藥材中分離得到的鳶尾甲黃素B、鳶尾黃素、次野鳶尾黃素及野鳶尾黃素對脂多糖誘導的小鼠巨噬細胞產生的一氧化氮均有抑制作用[7]。射干苷、野鳶尾苷的抗炎作用雖然不如其相應的苷元,但胃腸吸收動力學試驗表明,射干苷、野鳶尾苷在體內可以轉化為相應苷元而發揮藥理作用[8]。
2015年版《中華人民共和國藥典》射干項下僅對次野鳶尾黃素的含量進行了測定[9],目前亦有報道射干藥材中多種成分的含量測定方法[10-12]。本試驗同時對射干中9種活性成分含量進行測定,為進一步完善射干藥材的質量控制方法提供參考。
1 儀器與試藥
Agilent 1100高效液相色譜儀,配備在線脫氣機、四元梯度泵、自動進樣器、柱溫箱、DAD檢測器、Chemstation色譜工作站,美國Agilent公司;KQ-250 DB數控超聲波清洗器,昆山市超聲儀器有限公司。
對照品芒果苷(批號111607-200402)、射干苷(批號111632-200501)、次野鳶尾黃素(批號111557-200602)購于中國食品藥品檢定研究院,野鳶尾苷(批號13030803)、野鳶尾黃素(批號為13051402)購于成都普瑞法科技有限公司,鳶尾黃素(批號12081808)購于成都生物科技有限公司,鳶尾甲黃素A(批號20140724)、鳶尾甲黃素B(批號20140616)、白射干素(批號20140621)購于江蘇永健醫藥科技有限公司。甲酸、乙腈、DMSO均為色譜純(Dikma Technologies Inc.),重蒸水(自制)。所購射干藥材經遼寧中醫藥大學李峰教授鑒定,均為鳶尾科射干屬植物射干Belamcanda chinensis(L.)DC.的干燥根莖,其來源及鑒定結果見表1。

表1 射干藥材來源及鑒定結果
2 方法與結果
2.1 溶液的制備
2.1.1 混合對照品貯備液 精密稱取對照品芒果苷、射干苷、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、白射干素及次野鳶尾黃素適量,用DMSO分別配制成濃度為2.140、4.370、4.600、0.784 0、1.380、1.020、2.260、0.516 3、1.510 g/L的對照品貯備液。精密量取鳶尾甲黃素A及野鳶尾黃素各0.5 mL,其余7種對照品各1 mL,置于同一10 mL量瓶中,用DMSO稀釋至刻度,搖勻,即得混合對照品貯備液(芒果苷214.0 mg/L、射干苷437.0 mg/L、野鳶尾苷460.0 mg/L、鳶尾黃素78.40 mg/L、鳶尾甲黃素B 138.0 mg/L、鳶尾甲黃素A 51.00 mg/L、野鳶尾黃素113.0 mg/L、白射干素51.63 mg/L、次野鳶尾黃素151.0 mg/L)。
2.1.2 供試品溶液 取射干藥材粉末(過40目篩)約1 g,精密稱定。置100 mL具塞錐形瓶中,精密加入70%乙醇25 mL,密塞,稱定質量,超聲提取(250 W,50 kHz)30 min,冷卻,稱定質量,用70%乙醇補足減失的質量,搖勻,濾過,用0.22 μm微孔濾膜過濾,取續濾液作為供試溶液。
2.2 色譜條件
色譜柱:LeapsilTMC18(100 mm×2.1 mm,3 μm);流動相:0.1%甲酸水溶液(A)和乙腈(B),梯度洗脫(0~10 min,95%~90%A;10~15 min,90%~86%A;15~90 min,86%~86%A;90~95min,86%~81%A;95~155 min,81%~81%A;155~160 min,81%~5%A);流速0.5 mL/min;柱溫:40 ℃;程序進樣:吸取供試液5~20 μL,然后吸取水(含10% DMSO)90 μL,混合10次,等待0.2 min后進樣分析。在上述色譜條件下,芒果苷、射干苷、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、白射干素及次野鳶尾黃素與相鄰色譜峰的分離度均符合要求,理論塔板數按野鳶尾黃素峰計算不低于6000,色譜圖見圖1。
2.3 線性關系考察
分別精密量取混合對照品貯備液1.0、2.0、4.0、8.0、10、12 μL,進樣分析,以溶液的進樣質量為橫坐標,以峰面積(A)為縱坐標,進行線性回歸,分別得到射干中9種成分的回歸方程、相關系數及線性范圍,見表2。
2.4 精密度試驗
精密吸取同一混合對照品溶液,連續進樣6次,程序進樣,測得芒果苷、射干苷、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、白射干素及次野鳶尾黃素峰面積的RSD分別為1.2%、2.8%、1.3%、0.3%、2.1%、1.2%、2.0%、2.4%、1.4%,表明精密度良好。
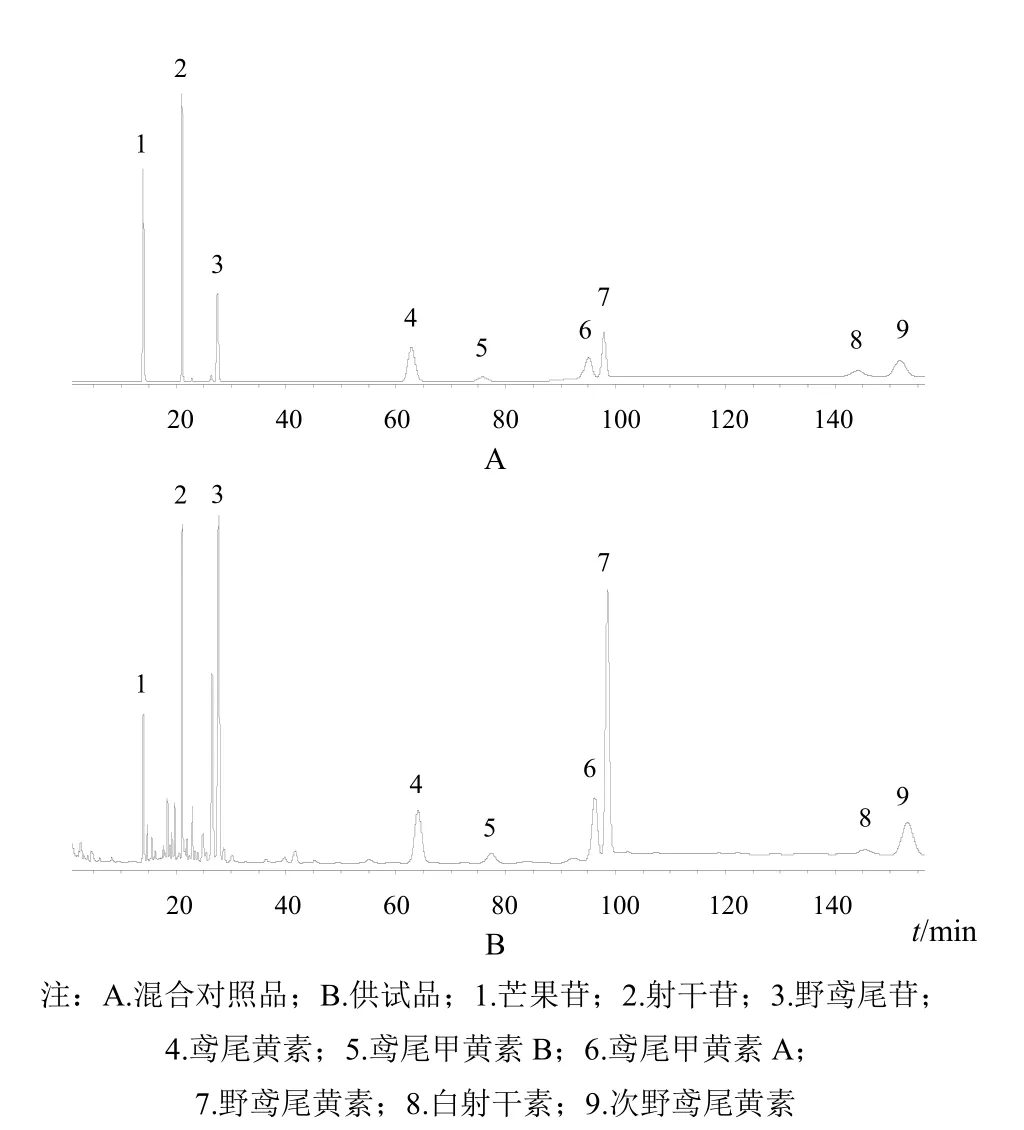
圖1 射干中9種活性成分HPLC圖
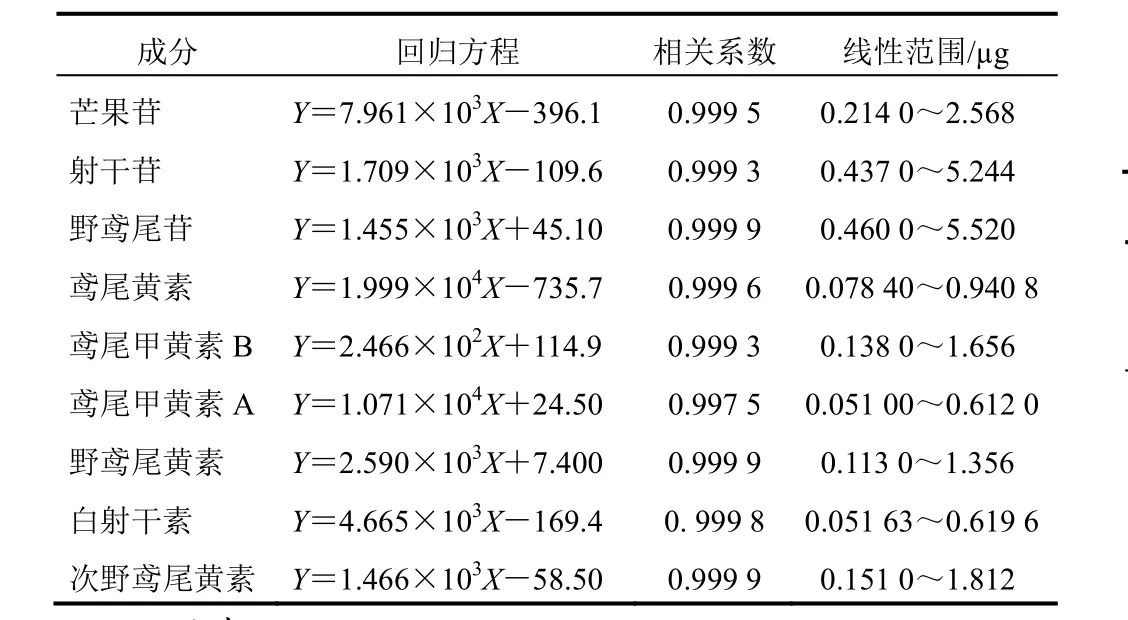
表2 射干中9種成分線性關系考察結果
2.5 重復性試驗
取同一批射干藥材粉末(1號),按“2.1.2”項下方法平行制備供試品溶液6份,在“2.2”項色譜條件下分別進樣分析,測得芒果苷、射干苷、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、白射干素及次野鳶尾黃素含量分別為4.17、1.90、1.356、1.639、0.211、0.974、1.042、1.197、0.859 mg/g,RSD分別為2.2%、2.6%、2.8%、2.1%、2.9%、2.7%、2.7%、2.5%、2.9%,表明方法重復性良好。
2.6 穩定性試驗
取新制備的供試品溶液,室溫放置,分別于0、3、6、9、24、96 h程序進樣,芒果苷、射干苷、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、白射干素及次野鳶尾黃素含量的RSD分別為0.18%、0.25%、0.21%、0.24%、0.23%、1.4%、0.25%、2.8%、2.6%,表明供試品溶液室溫放置96 h內穩定。
2.7 加樣回收率試驗
精密稱取已測知含量的射干藥材粉末(1號)9份,每份約0.5 g,精密稱定,分別精密加入含9種對照品的混合溶液1.2、1.0、0.8 mL(芒果苷2.099 g/L、射干苷0.927 0 g/L、野鳶尾苷0.688 0 g/L、鳶尾黃素0.839 0 g/L、鳶尾甲黃素B 0.107 0 g/L、鳶尾甲黃素A 0.489 0 g/L、野鳶尾黃素0.524 0 g/L、白射干素0.599 0 g/L、次野鳶尾黃素0.432 0 g/L),每一質量濃度制備3份。按“2.1.2”項下方法制備所需溶液,分別進樣分析,結果見表3~表11。
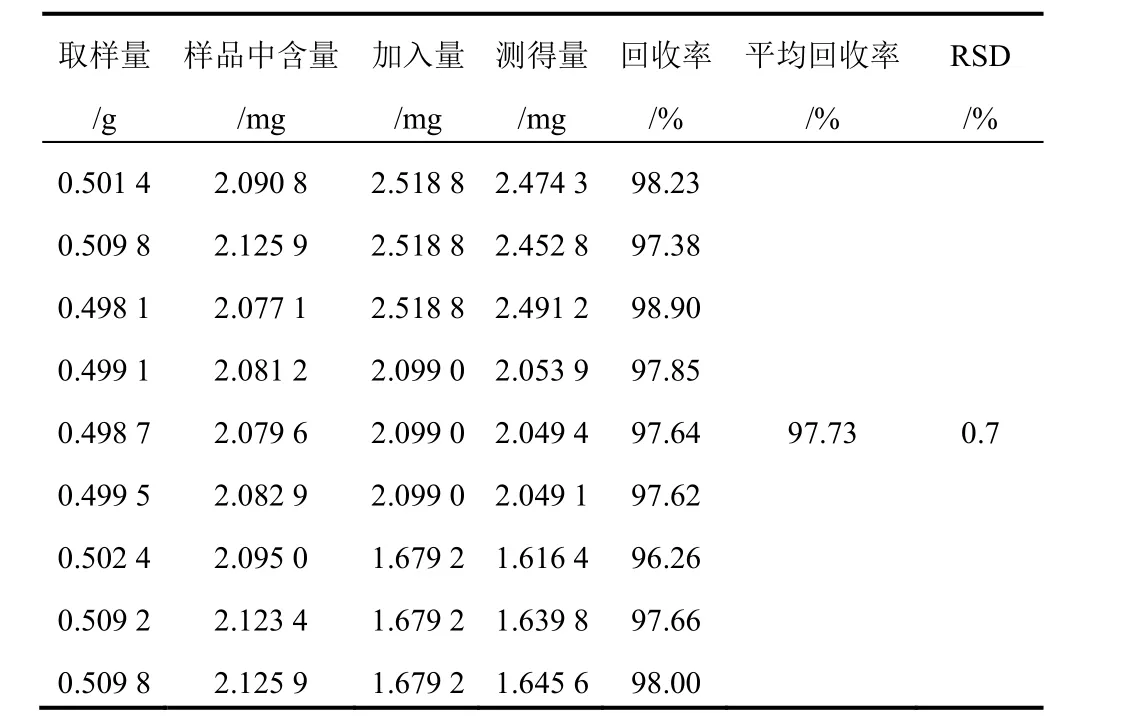
表3 射干中芒果苷加樣回收率試驗
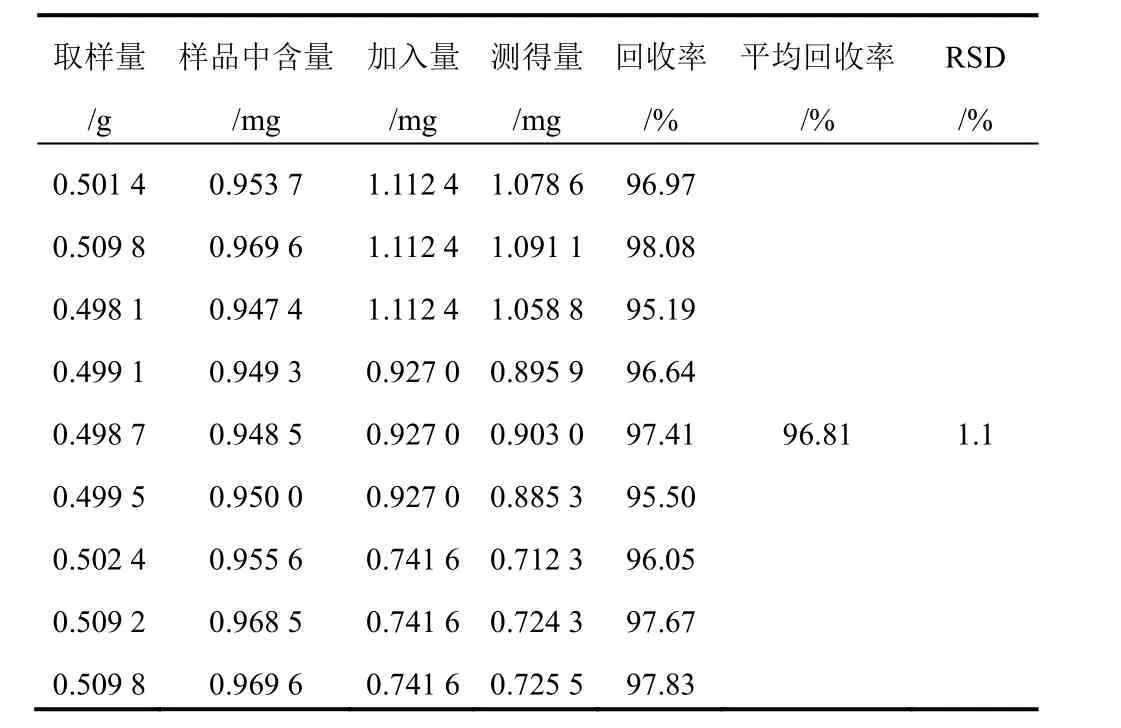
表4 射干中射干苷加樣回收率試驗
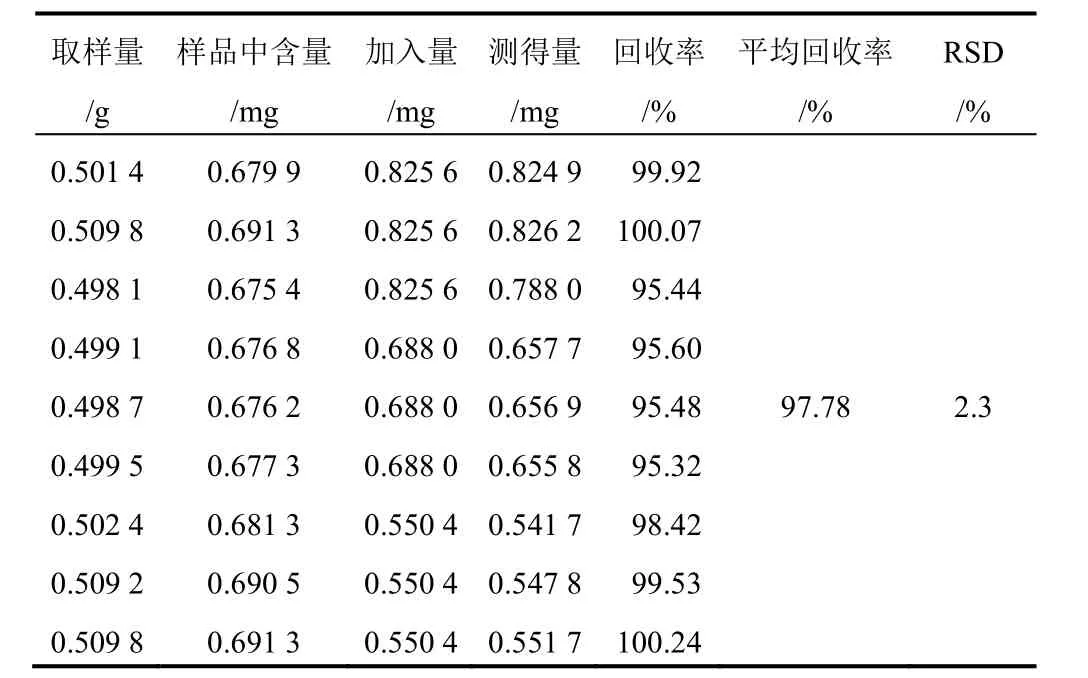
表5 射干中野鳶尾苷加樣回收率試驗
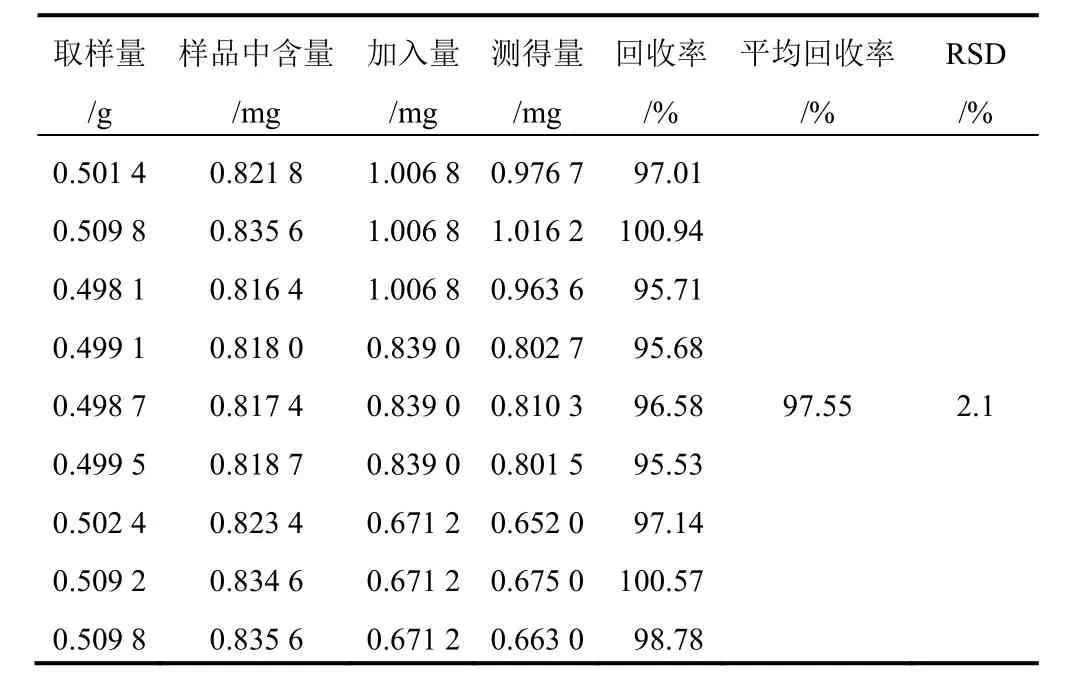
表6 射干中鳶尾黃素加樣回收率試驗

表7 射干中鳶尾甲黃素B加樣回收率試驗
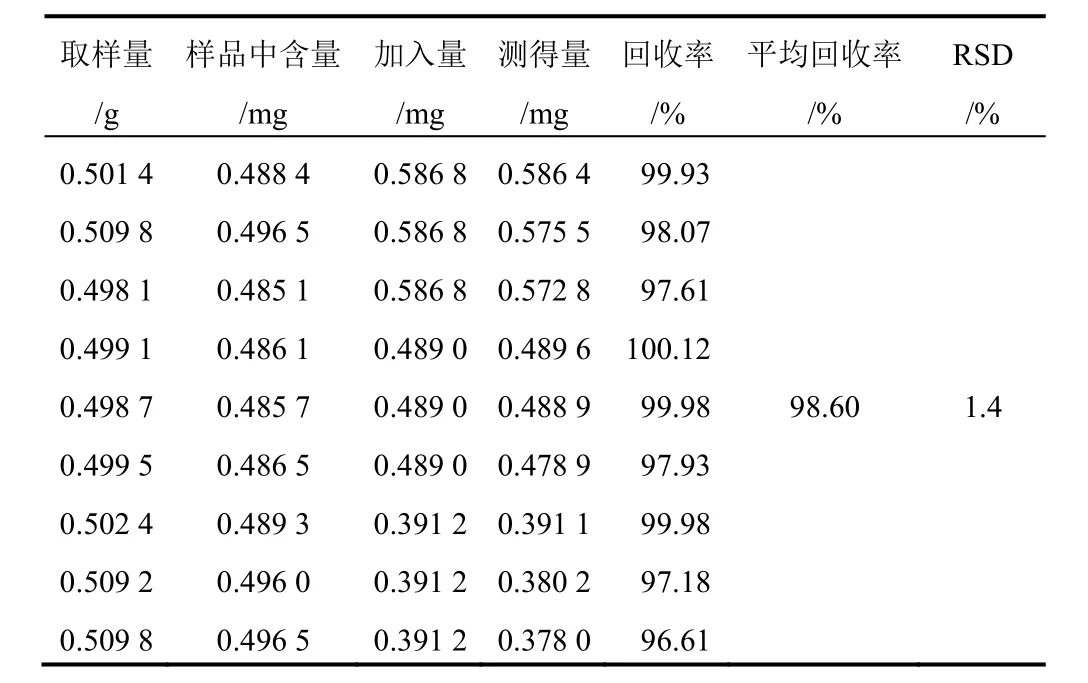
表8 射干中鳶尾甲黃素A加樣回收率試驗
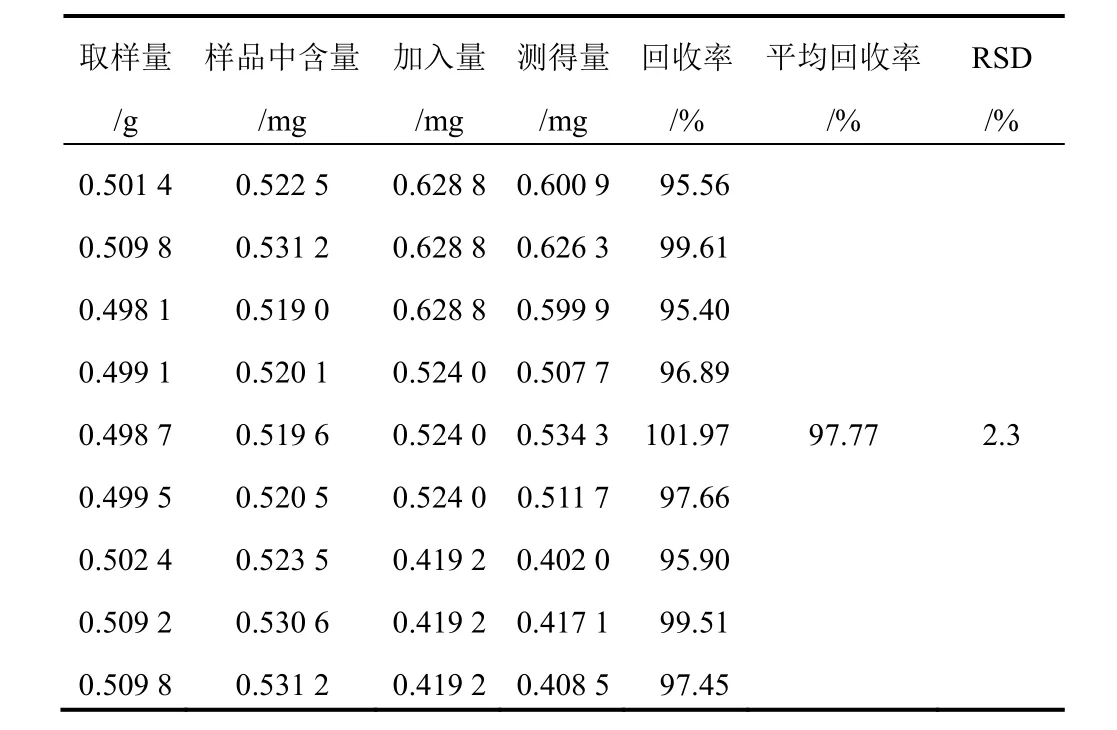
表9 射干中野鳶尾黃素加樣回收率試驗
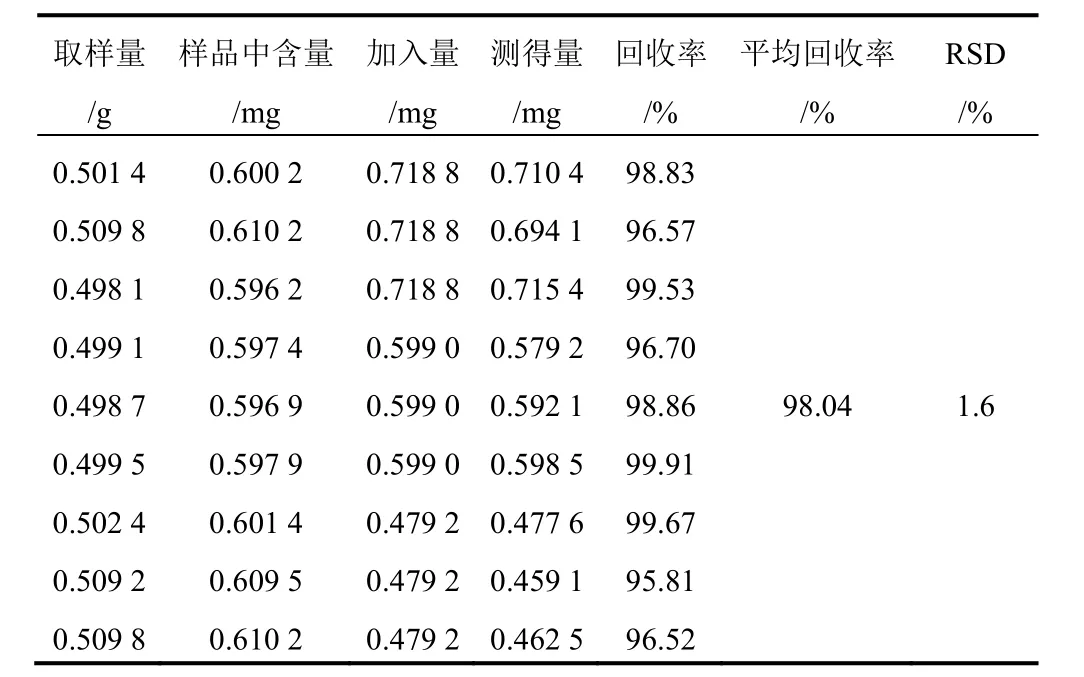
表10 射干中白射干素加樣回收率試驗

表11 射干中次野鳶尾黃素加樣回收率試驗
2.8 樣品含量測定
取不同產地的5批射干藥材粉末,按“2.1.2”項下方法制備供試品溶液,在“2.2”項色譜條件下進行分析,用外標法計算芒果苷、射干苷、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、白射干素及次野鳶尾黃素的含量,結果見表12。
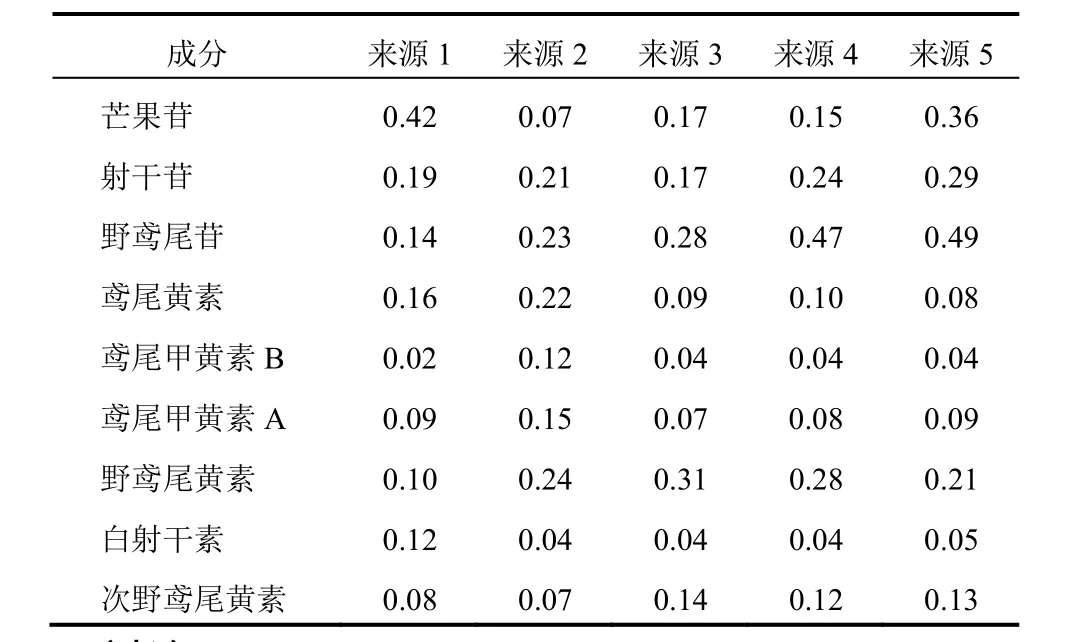
表12 不同來源射干藥材中9種活性成分含量(%)
3 討論
射干所含9種成分中除芒果苷外,其他均為異黃酮成分,化學結構相近,較難分離,其中鳶尾甲黃素A與野鳶尾黃素結構僅差一個甲氧基,色譜行為非常接近,色譜峰很難分開。本研究從色譜柱粒徑、流動相比例、柱溫、流速、檢測波長等多方面進行考察,對色譜條件進行優化,并采用程序進樣對供試品溶液的離子強度與流動相進行匹配,使各成分色譜峰得到良好分離,最終實現了9個色譜峰的準確測定。
選擇色譜柱時曾采用Agilent ZORbax SB-Aq、Welch Ultimate XB-C18、Phenomenex Polar-BP等多種填料的色譜柱進行分離,除鳶尾甲黃素A與野鳶尾黃素不能分開外,其他7種成分分離較好。當柱粒徑為5 μm、色譜柱內徑為4.6 mm時,調整色譜柱類型、柱溫、流動相種類及比例均無法使鳶尾甲黃素A與野鳶尾黃素2個峰分開。當柱粒徑為3 μm、色譜柱內徑為2.1 mm時,分離效果良好,因此確定采用LeapsilTMC18色譜柱(100 mm×2.1 mm,3 μm)。
在LeapsilTMC18色譜柱(100 mm×2.1 mm,2.7 μm)柱上,野鳶尾黃素與鳶尾甲黃素A、白射干素與次野鳶尾黃素分離度較差;在經過多次試驗后發現,當乙腈的體積分數為14%,且在0.5 mL/min運行75 min左右,野鳶尾黃素與鳶尾甲黃素A分離度可以達到定量要求;當乙腈體積分數為19%且在0.5 mL/min運行60 min左右,次野鳶尾黃素與白射干素色譜峰分離度可以達到定量要求;再考慮極性較大的芒果苷、射干苷、野鳶尾苷等分離需要,最終確定了最佳流動相梯度。
由于色譜柱的內徑及粒徑均較小,1.0 mL/min流速使柱壓過高,升高柱溫和降低流速既降低柱壓又可獲得良好的分離效果,因此確定流速為0.5 mL/min,柱溫為40 ℃。
由于同時進行測定的9個成分的極性差異較大,經過篩選,采用70%乙醇可以使各成分得到良好的溶出,因此確定用70%乙醇為提取溶媒,但芒果苷、射干苷及野鳶尾苷的色譜峰由于供試品溶液溶劑離子強度遠大于分離時流動相離子強度,因此色譜峰變形,難以準確積分,經過反復試驗,將供試品溶液用水稀釋20倍左右,才能獲得良好結果,但會使進樣量加大,同樣導致色譜峰變形,采用Agilent1100液相色譜儀提供的程序進樣功能,可以較好地解決這個問題。另外,由于樣品中的射干異黃酮苷元的脂溶性較強,直接用水稀釋會使樣品中這些成分的含量降低,因此,在水中加入了一定比例的DMSO,最終確定了最佳進樣程序參數。
樣品測定結果表明,本研究所收集到的5批樣品中,芒果苷含量為0.07%~0.42%,射干苷含量為0.17%~0.29%,野鳶尾苷含量為0.14%~0.49%,鳶尾黃素含量為0.08%~0.22%,鳶尾甲黃素B含量為0.02%~0.12%,鳶尾甲黃素A含量為0.07%~0.15%,野鳶尾黃素含量為0.10%~0.31%,白射干素含量為0.04%~0.12%,次野鳶尾黃素含量為0.07%~0.14%。從生長環境看,安徽野生射干中芒果苷和白射干素含量比栽培高,其他成分含量比栽培低,而遼寧野生射干除芒果苷略低于栽培外,其他8種成分含量非常接近,3個產地的射干中各成分含量規律性不強,可能是樣品批次較少的原因。
[1] 馮超.射干異黃酮類成分質量控制方法研究[D].上海:第二軍醫大學, 2009.
[2] 李國信,秦文艷,齊越,等.射干提取物抗炎及鎮痛藥理實驗研究[J].實用中醫內科雜志,2008,22(1):3-4.
[3] 秦文艷,趙金明,齊越,等.射干提取物體內體外抑菌作用的研究[J].中國實驗方劑學雜志,2011,17(4):147-150.
[4] 趙金明,孟莉,陳賀,等.射干有效成分抗病毒主要藥效學實驗研究[J].實驗動物科學,2010,27(6):9-12.
[5] KIM Y P, YAMADA M, LIM S S, et al. Inhibition by tectorigenin and tectoridin of prostaglandin E2production and cyclooxygenase-2 induction in rat peritoneal macrophages[J]. Biochim Biophys Acta, 1999,1438(3):399-407.
[6] AHNA K S, NOBE E J, CHAC K H. et al. Inhibitory effects of Irigenin from the rhizomes of Belamcanda chinensis on nitric oxide and prostaglandin E2production in murinemacrophage RAW 264.7 cells[J]. J Life Sciences,2006,78:2336-2342.
[7] LEE J W, LEE C, JIN Q, et al. Chemical constituents from Belamcanda chinensis and their inhibitory effects on nitric oxide production in RAW 264.7 macrophage cells[J]. Arch Pharm Res,2015, 38(6):991-997.
[8] BAE E A, HAN M J, LEE K T, et al. Metabolism of 6”-O-xylosyltectoridin and tectoridin by human intestinal bacteria and their hypoglycemic and in vitro cytotoxic activities[J]. Biol & Pharm Bull,1999,22(12):1314-1318.
[9] 國家藥典委員會.中華人民共和國藥典:一部[M].北京:中國醫藥科技出版社,2015:285.
[10] 張婧涵,張曉瑞,李國信,等.線性回歸色譜峰定位法在射干中藥材多組分同時測定中的應用[J].藥物分析雜志,2014,34(7):1149-1154.
[11] 鄒桂欣,尤獻民,李國信.射干中芒果苷反相高效液相色譜測定[J].遼寧中醫雜志,2010,37(10):2002-2003.
[12] 鄒桂欣,尤獻民,李國信.HPLC測定射干不同部位中的4種藥用成分[J].華西藥學雜志,2011,26(2):170-171.
Simultaneous Determination of Nine Active Ingredients in Belamcandae Rhizoma by RP-HPLC
YOU Xian-min, ZOU Gui-xin, DI Zi-zhen, LI Guo-xin (Liaoning Academy of Chinese Medicine,
Shenyang 110034, China)
Objective To develop an RP-HPLC method for simultaneous determination of mangiferin, tectoridin, iridin, tectorigenin, iristectorigenin B, iristectorigenin A, irigenin, dichotomin and irisflorentin in Belamcandae Rhizoma. Methods Separation was carried out on an LeapsilTMC18 column (100 mm×2.1 mm, 3 μm) with an isocratic mobile phase consisting of acetonotrile and formic acid at a flow rate of 0.5 mL/min; The detection wavelength was set at 265 nm; the column temperature was 40 ℃. Results The linear ranges of mangiferin, tectoridin, iridin, tectorigenin, iristectorigenin B, iristectorigenin A, irigenin, dichotomin and irisflorentin were 0.214 0–2.568 μg (r=0.999 5), 0.437 0–5.244 μg (r=0.999 3), 0.460 0–5.520 μg (r=0.999 9), 0.078 40–0.940 8 μg (r=0.999 6), 0.138 0–1.656 μg (r=0.999 3), 0.051 00–0.612 0 μg (r=0.997 5), 0.113 0–1.356 μg (r=0.999 9), 0.051 63–0.619 6 μg (r=0.999 8) and 0.151 0–1.812 μg (r=0.999 9), respectively. The average recoveries were 97.73%, 96.81%, 97.78%, 97.55%, 96.86%, 98.60%, 97.77%, 98.04% and 97.89%, respectively; the relative standard deviations were 0.70%, 1.1%, 2.3%, 2.1%, 1.3%, 1.4%, 2.3%, 1.6% and 1.9%, respectively. This method was used to determine the contents of nine active ingrients in 5 batches of Belamcandae Rhizoma. Conclusion The method is accurate and reliable, which can be used for the quality control of Belamcandae Rhizoma.
Belamcandae Rhizoma; active ingredients; RP-HPLC; content determination
10.3969/j.issn.1005-5304.2017.01.020
R284.1
A
1005-5304(2017)01-0082-05
2016-02-13)
(
2016-03-09;編輯:陳靜)
國家自然科學基金(81273927);國家臨床重點專科建設項目(2013年);國家中醫藥管理局臨床中藥學重點學科(2009年);遼寧中醫藥大學杏林學者青藍工程(2013年)
李國信,E-mail:zou650430@126.com
