JAG1基因突變Alagille綜合征1例報(bào)告并文獻(xiàn)復(fù)習(xí)
2017-12-28 02:31:05賴慧儀戴煒朱質(zhì)斌
中西醫(yī)結(jié)合肝病雜志 2017年5期
賴慧儀 戴煒 朱質(zhì)斌
深圳市第三人民醫(yī)院肝四科 (廣東 深圳,518000)
·病例報(bào)告·
JAG1基因突變Alagille綜合征1例報(bào)告并文獻(xiàn)復(fù)習(xí)
賴慧儀 戴煒 朱質(zhì)斌
深圳市第三人民醫(yī)院肝四科 (廣東 深圳,518000)
Alagille綜合征是一種累及多系統(tǒng)的常染色體顯性遺傳性疾病。該綜合征在1969年由Alagille等首次報(bào)道,并在1975年得到進(jìn)一步闡述。Alagille綜合征涉及的臟器包括肝臟、心臟、骨骼、眼睛和顏面等,國外報(bào)道該病的發(fā)病率約為1/70000[1]。國內(nèi)少有報(bào)道。現(xiàn)將我們收治的1例JAG1基因突變Alagille綜合征致肝硬化失代償病例報(bào)告如下。
1 病例 患者,女,20歲。因反復(fù)皮膚黃染20年入院,患者1996年出生后不久即出現(xiàn)身目黃染,伴白陶土樣大便,當(dāng)時(shí)疑似診斷“先天性膽道閉鎖”。1999年(3歲)查肝功能:ALT 126U/L,AST 206U/L,TBil 164.8μmol/l,DBil 106.5μmo/l,GGT 408U/L,CMV-IgM陽性、CMV DNA陽性,診斷“巨細(xì)胞病毒肝炎”,予更昔洛韋抗病毒治療,效果不明顯。2000年(4歲)行ECT檢查顯示:未見膽道畸形征象。2009年(13歲)行肝穿活檢術(shù):膽汁淤積性肝硬化,中度活動(dòng)并膽管缺乏,診斷“原發(fā)性膽汁性肝硬化”,予優(yōu)思弗治療。2011年(15歲)后反復(fù)因“肝硬化失代償”在我院住院,疑似診斷Alagille綜合征。2016年4月(20歲)再次住院。母親懷孕期間無感染或接觸藥物、毒物、射線史,家族無相關(guān)遺傳病史,出生后體格及智力發(fā)育遲緩。查體:發(fā)育不良,身材矮小,面色晦暗,皮膚、鞏膜輕度黃染,肝掌、蜘蛛痣(+),頭顱偏小,前額突出,眼與鼻的距離偏大,眼科檢查可見可疑“角膜后胚胎環(huán)”(見圖1),K-F環(huán)陰性,存在先天性眼震和黃斑發(fā)育異常。下頦小而尖,耳廓突出,牙齒色素沉著并多個(gè)牙齒斷裂脫落(見圖2)。主動(dòng)脈瓣聽診區(qū)可聞及III級收縮期雜音。肝右肋下2cm,劍突下5cm可及,質(zhì)硬,邊鈍,表面光滑,無觸痛,脾平臍。查血常規(guī):WBC 0.46×109/L,N 0.19×109/L,RBC 2.32×1012/L,HGB 73g/L,PLT 18×109/L;凝血功能:PT 18.7秒,PTA 54%;肝功能:Alb 20.6g/L,TBil 40.4 μmol/L,DBil30.8μmol/L,ALT 92U/L,AST 273U/L,GGT 218U/L,ALP 277U/L;血脂:LDL 3.44mmol/L。嗜肝病毒肝炎系列:HAV-IgG陽性,HAV-IgM陰性,乙型、丙型、戊型肝類病毒標(biāo)志物全陰性;TORCH、EBV-IgM陰性;ANA、AMA陰性。血清銅蘭蛋白、血清銅正常。血糖、血清鐵及α1抗胰蛋白酶正常。心臟彩超:二尖瓣、主動(dòng)脈瓣輕度返流。頸椎、胸椎DR:C2/3椎間隙稍窄,胸10椎體裂椎畸形(蝴蝶椎)(見圖3)。上腹部CTA:肝臟形態(tài)改變,脾動(dòng)脈多發(fā)瘤樣擴(kuò)張;肝硬化,門脈高壓,巨脾。單基因遺傳病基因檢測報(bào)告(檢測方法:芯片捕獲高通量測序方法獲知缺失區(qū)域,qPCR方法對缺失區(qū)域進(jìn)行驗(yàn)證):JAG1基因的疑似致病突變EX1_26DEL(Het),經(jīng)過基因檢測驗(yàn)證報(bào)告確定該基因突變存在。

圖1 箭頭處角膜后胚胎環(huán)
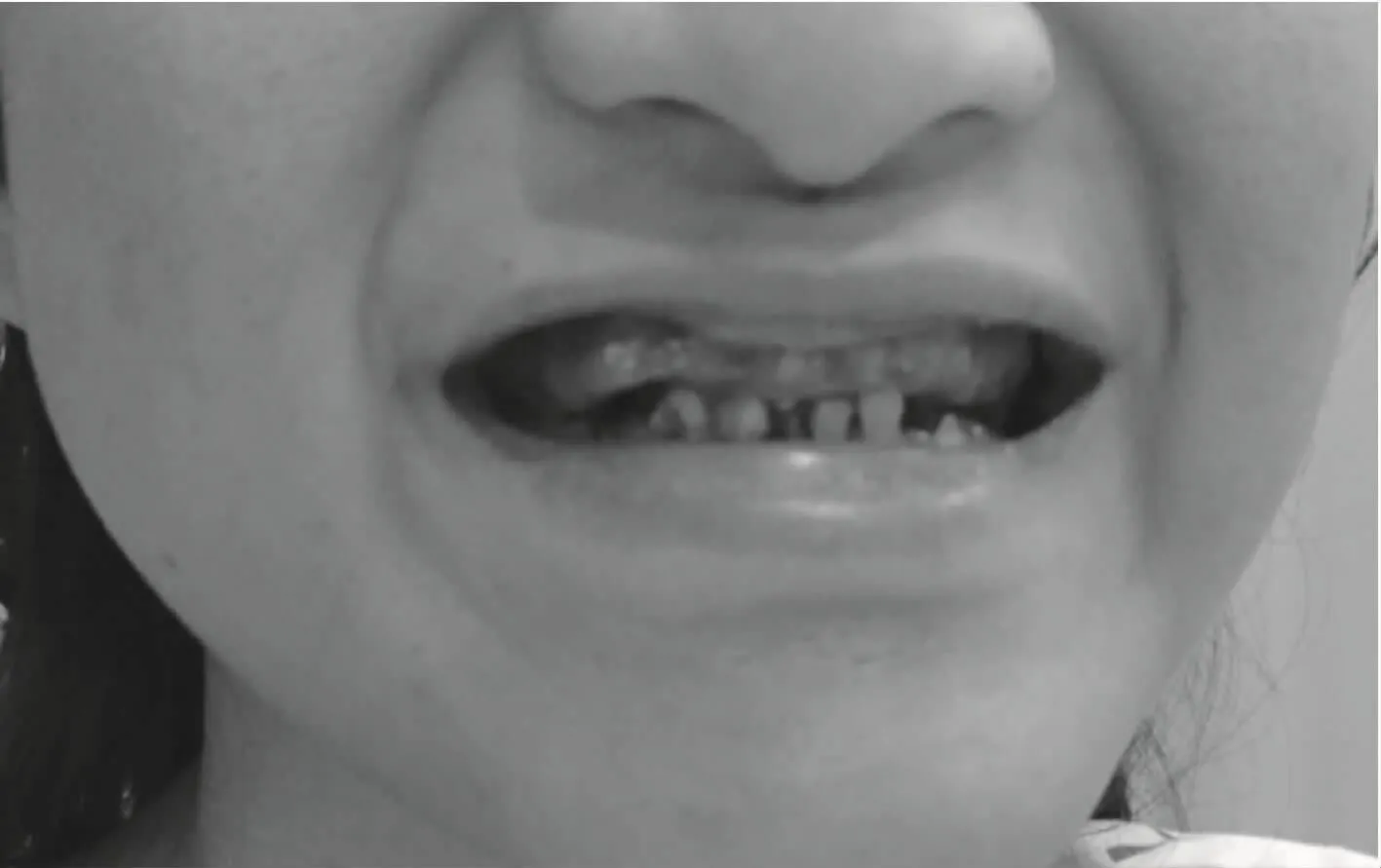
圖2 牙齒色素沉著并多個(gè)牙齒斷裂脫落
2 討論
Alagille綜合證(ALGS)又稱先天性肝內(nèi)膽管發(fā)育不良征,是一種累及多系統(tǒng)的顯性遺傳性疾病。臨床表現(xiàn)為膽汁淤積伴瘙癢和小葉間膽管減少,先天性心臟病、輕微椎體分割異常、特征性面容、角膜后胚胎環(huán),以及腎臟發(fā)育不良等[1]。病因是JAG1基因(20p12)或者NOTCH2基因(1p13)突變導(dǎo)致共同Notch信號通路缺陷,約90%的ALGS由編碼JAGGED1的JAG1基因突變或缺失所引起,導(dǎo)致1型ALGS,常符合經(jīng)典ALGS臨床特征,約1% 由NOTCH2基因突變導(dǎo)致2型ALGS,常有腎臟病變及膽管稀疏,而較少出現(xiàn)骨骼畸形及面部特征,不完全符合傳統(tǒng)診斷標(biāo)準(zhǔn)[2~4]。JAG1基因突變所致1型ALGS常為新發(fā)突變。該病例家族中無類似病史,考慮其基因突變?yōu)樾掳l(fā)突變。
ALGS患者慢性膽汁淤積發(fā)生率高達(dá)95%,在出生后3個(gè)月內(nèi)甚至新生兒期大部分患兒即開始出現(xiàn)膽汁淤積并逐漸發(fā)展,出現(xiàn)黃疸、皮膚瘙癢、白陶土樣大便及高脂血癥,常有肝脾腫大,膽紅素隨年齡增長有下降趨勢,很少發(fā)生肝硬化。肝臟病理表現(xiàn)為小葉間膽管缺乏。心血管異常(90%)表現(xiàn)常見肺動(dòng)脈狹窄,肺動(dòng)脈病變多單發(fā),也可與其他心臟病變同時(shí)出現(xiàn),其中周圍肺動(dòng)脈和肺動(dòng)脈瓣狹窄占67%,法洛四聯(lián)癥占16%[5-6]。ALGS患者出現(xiàn)蝴蝶椎的發(fā)生率占80%[7],骨骼異常通常無明顯癥狀,一般在X線檢查時(shí)發(fā)現(xiàn),發(fā)生原因是骨質(zhì)發(fā)育障礙和骨質(zhì)流失,具體表現(xiàn)為脊柱畸形,偶爾出現(xiàn)椎體融合、隱性脊柱裂等,除脊柱病變外,少數(shù)患者表現(xiàn)為四肢骨骼病變。特殊面容占95%[8]表現(xiàn)為前額寬闊,眼睛深陷,眼距增寬,有時(shí)可見瞼裂上斜、耳廓突出、鼻梁挺直而鼻尖呈蒜頭狀、尖下巴,上述特點(diǎn)使得臉呈倒三角形。眼部異常可見于90%的ALGS患者中,以角膜后胚胎環(huán)最常見,多發(fā)生于角膜內(nèi)皮和葡萄膜(虹膜)小梁網(wǎng)[9],但這種角膜后胚胎環(huán)可發(fā)生在15%正常人群中,70%發(fā)生在22q11缺失綜合征的患者中[10]。此外,眼部異常還可影響到角膜、結(jié)膜、視網(wǎng)膜、視神經(jīng)盤等,可出現(xiàn)視網(wǎng)膜萎縮、黃斑發(fā)育不良,角膜鞏膜發(fā)育不全等,因此各種眼科癥狀都有可能發(fā)生。其他表現(xiàn)包括腎臟病變(40%),生長發(fā)育遲緩(50%),胰腺功能不全等,而口腔問題常常依賴于肝臟的疾病狀態(tài),由于膽汁長期淤積導(dǎo)致牙釉質(zhì)混濁、礦質(zhì)過少和牙齒的色素沉著等病變。[11,12]
經(jīng)典的ALGS診斷標(biāo)準(zhǔn):慢性膽汁淤積、心臟疾病、骨骼異常、眼部異常和面部特征等五條標(biāo)準(zhǔn)滿足三條即可診斷[13],具體見表1。結(jié)合家族史、病理、基因檢測,Kamath等[14]和 Guru等[15]提出了修訂的 AGLS診斷標(biāo)準(zhǔn)(見表1)。隨著基因診斷的進(jìn)展,越來越多患者可通過基因檢測來明確診斷。

表1 修訂的ALGS診斷標(biāo)準(zhǔn)
治療:目前尚無病因治療措施,以對癥支持治療為主,加強(qiáng)營養(yǎng)有利于生長發(fā)育改善。其最終預(yù)后取決于肝臟和心臟疾病的嚴(yán)重程度。若出現(xiàn)肝硬化失代償可考慮肝移植。Lee等[16]對9例ALGS兒童進(jìn)行肝移植手術(shù),活體肝移植后的5年期和20年總生存率分別為88.9%和77.8%。
該患者自出生開始發(fā)病,呈典型臨床表現(xiàn),基因檢測出JAG1基因突變,診斷JAG1基因突變致Alagille綜合征明確。因該病為常染色體顯性遺傳病,但患者家族中無相關(guān)病史,考慮為妊娠過程中胎兒基因產(chǎn)生突變導(dǎo)致發(fā)病。該病發(fā)病率極低,亦因基因檢測條件所限,診斷率極低,國內(nèi)報(bào)道也較少,造成就診過程中多次誤診。因此,兒科醫(yī)生及肝病醫(yī)生應(yīng)予重視。
[1]中華醫(yī)學(xué)會肝病學(xué)分會,中華醫(yī)學(xué)會消化病學(xué)分會、中華醫(yī)學(xué)會感染病學(xué)分會.膽汁淤積性肝病診斷和治療共識(2015)[J].胃腸病學(xué),2016,21(1):39-49.
[2]Leonard LD,Chao G,Baker A,et al.Clinical utility gene card for:Alagille syndrome(ALGS)[J].Eur JHum Genet,2014,22(3):1-4.
[3]Vozzi D,Licastro D,Martelossi S,et al.Alagille syndrome:a new missensemutation detected bywhole-exome sequencing in a case previously found to be negative by DHPLC and MLPA[J].Mol Syndromol,2013,4(4):207-210.
[4]Kamath BM,Bauer RC,Loomes KM,et al.NOTCH2 mutations in Alagille syndrome[J].JMed Genet,2012,49(2):138-144.
[5]Emerick KM,Rand EB,Goldmuntz E,etal.Features of Alagille syndrome in 92 patients:frequency and relation to prognosis[J].Hepatology,1999,29(5):822-829.
[6]McElhinney DB,Krantz ID,Bason Let al.Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome[J].Circulation,2002,106(7):2567-2574.
[7]Sanderson E,Newman V,Haigh SF,et al.Vertebral anomalies in children with Alagille syndrome:an analysis of50 consecutive patients[J].Pediatr Radiol,2002,32(3):114-119.
[8]Alagille D,Estrada A,HadchouelM,etal.Syndromic paucity of interlobular bile ducts(Alagille syndrome or arteriohepatic dysplasia):review of 80 cases[J].JPediatr,1987,110(2):195-200.
[9]Mozhgan Z,Bita G,Mahmood H,et al.Paucity of intrahepatic bile ducts in neonates:the first case series from iran[J].Iran JPediatr,2013,23(1):65-70.
[10]McDonald-McGinn DM,Kirschner R,Goldmuntz E,et al.The Philadelphia story:the 22q11.2 deletion:reporton 250 patients[J].Genet Couns,1999,10(1):11-24.
[11]Olczak-Kowalczyk D,Paw lowska J,Kowalczyk W.Oral health status in children with chronic liver disease[J].J Stoma,2011,64(10):760-774.
[12]Berniczei-Royko A,Chalas R,Mitura I,et al.Medical and dental management of Alagille syndrome:a review[J].Med Sci Monit,2014,20:476-480.
[13]Witt H,Neumann LM,Grollmuss O,et al.Prenatal diagnosis of Alagille syndrome[J].J Pediatr Gastroenterol Nutr,2004,38(1):105-106.
[14]Kamath BM.Alagille syndrome[M].In:Suchy FJ,Sokal RJ,Balistreri WF,editors Liver Disease in Children.3rd ed.New York:Cambridge University Press,2007:326-345.
[15]Guru Murthy GS,Rana BS,Das A,et al.Alagille syndrome:a rare disease in an adolescent[J].Dig Dis Sci,2012,57(11):3035-3057.
[16]Lee CN,Tiao MM,Chen HJ,et al.Characteristics and outcome of liver transplantation in children with Alagille syndrome:a single-center experience[J].Pediatr Neonatol,2014,55(2):135-138.
10.3969/j.issn.1005-0264.2017.05.019
2017-03-24 編輯:吳輝坤)
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學(xué)生導(dǎo)刊(2018年13期)2018-06-29 03:49:00
現(xiàn)代檢驗(yàn)醫(yī)學(xué)雜志(2016年4期)2016-11-15 02:01:14
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
