基于兩種一測多評法對尾葉香茶菜中四種二萜類成分的質量控制研究
2018-02-22 10:39:30楊麗霞陳勇強
山西中醫藥大學學報 2018年6期
楊麗霞,陳勇強,孫 艦
(1.山西中醫藥大學,山西晉中030619; 2.晉中學院化學化工學院,山西晉中030619;3.廣西中醫藥大學賽思斯醫藥學院,廣西南寧530222)
尾葉香茶菜 (Rabdosia excisa(Maxim.)Hara)又名龜葉草,是唇形科香茶菜屬(Rabdosia)植物,在我國的東北、日本、朝鮮以及俄羅斯遠東地區都有分布[1],全草入藥,具有清熱解毒、健胃、活血的功效。用于治療胃炎、膀胱脹痛、感冒發熱、乳腺癌、關節痛、蛇蟲咬傷等[2]。藥理研究表明,尾葉香茶菜中的二萜類化合物Kamebakaurin、excisanin A、rabdokunmin C和Kamebanin對P388有明顯的抑制作用[3-4]。目前,用于尾葉香茶菜藥材質量控制與評價的方法主要是對尾葉香茶菜活性成分含量的精確測定[5-7],不能全面科學地控制與評價尾葉香茶菜藥材質量;同時,多個活性成分的含量測定需要耗費多種對照品,而一些對照品又存在難以制備、不穩定、難以獲得或價格昂貴等問題。一測多評法的應用可以解決對照品缺乏的難題。本實驗采用HPLC法同時測定了尾葉香茶菜中Kamebakaurin、excisanin A、rabdokunmin C和Kamebanin的含量,并利用以上4種二萜類物質內在的函數和比例關系,采用一測多評法[8-9],以 Kamebakaurin 為內參物,計算得 excisanin A、rabdokunmin C、Kamebanin與Kamebakaurin之間的相對校正因子(fk/s),從而得出4種二萜類物質的含量,為更確切評價尾葉香茶菜藥材的質量提供了新的分析模式。
1 材 料
1.1 儀器
LC-20A高效液相色譜儀,Diamonsil C18色譜柱(250 mm×4.6 mm,5 μm),Shim-pack VP-ODS C18色譜柱(250 mm×4.6 mm,5 μm),InertSustain C18色譜柱(250 mm×4.6 mm,5 μm)均為日本島津公司;DV215CD分析天平(十萬分之一,美國OHAUS公司);DV114C分析天平(萬分之一,美國OHAUS公司);KQ5200V超聲清洗儀(江蘇省昆山市淀山湖鎮)。
1.2 試 藥
8批尾葉香茶菜藥用植物采集于遼寧本溪、撫順、丹東、鞍山,并按照《中國藥典》2015版(一部)規定進行干燥,粉碎,過40目篩等預處理[10];對照品 Kamebakaurin、excisanin A、rabdokunmin C和Kamebanin由本實驗室制備,純度均在98%以上。乙腈(色譜純,天津四有生物醫學科技有限公司);甲醇(分析純,天津科密歐化學試劑有限公司);純凈水(杭州娃哈哈集團有限公司)。
2 方法與結果
2.1 HPLC定量方法
2.1.1 色譜條件 色譜柱:Diamonsil C18柱(250 mm×4.6 mm,5 μm);流動相為乙腈-水(體積比23∶77);流速 1.0 mL/min;運行時間 65 min;柱溫25℃;檢測波長230 nm;進樣量20 μL;兩次進樣之間用流動相平衡15 min。色譜圖見圖1。
2.1.2 混合對照品溶液的制備 精密稱取對照品Kamebakaurin 16.82 mg、excisaninA 5.85 mg、rabdokunmin C 4.82 mg和Kamebanin 4.38 mg分別置于25 mL容量瓶中,加甲醇溶解并稀釋至刻度。分別精密吸取上述溶液各1.0 mL置于同一10 mL容量瓶中,用甲醇定容,搖勻即得混合對照品溶液。
2.1.3 供試品溶液的制備 取干燥尾葉香茶菜藥材均勻粉末,精密稱取1.0 g,用甲醇提取3次(2 h、1.5 h、1 h),每次加甲醇 15 mL,水浴加熱回流溫度72℃,合并提取液,減壓過濾得浸膏,加甲醇溶解浸膏并定容至100 mL,搖勻,用0.45 μm微孔濾膜過濾,取其濾液,即得。
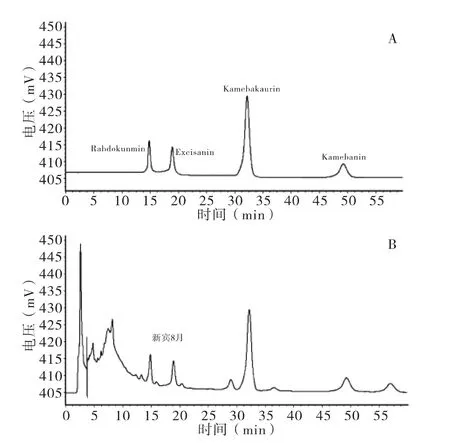
圖1 混合對照品(A)和供試品(B)HPLC圖
2.1.4 線性關系考察 精密稱量化合物Kamebakaurin、excisaninA、rabdokunmin C 和 Kamebanin的對照品,分別配制成濃度為0.005 mg/mL,0.015 mg/mL,0.03 mg/mL,0.06 mg/mL,0.12 mg/mL,0.24 mg/mL的單一對照品溶液,分別取上述對照品溶液20 μL 進樣分析,以進樣量(X,μg)對峰面積積分值(Y)進行回歸,得 Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin 的回歸方程,見表 1,各標準曲線在線性范圍(0.1~4.8 μg)內線性良好。

表1 4種成分的線性關系考察結果
2.1.5 精密度試驗 精密吸取同一混合對照品溶液20 μL,連續進樣6次,記錄各色譜峰峰面積,計 算 得 Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin峰面積的 RSD(n=6)分別為 0.73%、0.52%、0.44%、0.34%,表明儀器精密度良好。
2.1.6 穩定性試驗 取干燥尾葉香茶菜藥材均勻粉末(新賓8月)1.0 g,按“2.1.3”項制得供試品溶液,在0~12 h內每隔2 h進樣1次,每次進樣20 μL,測定峰面積。利用峰面積平均值考察樣品在甲醇溶液中的穩定性,結果顯示,化合物Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin 在各時間點峰面積的RSD(n=6)分別為0.94%、1.12%、1.18%、1.09%,說明樣品在甲醇中12 h內穩定性良好。
2.1.7 重復性試驗 取干燥尾葉香茶菜藥材均勻粉末(新賓8月)6份,每份 1.0 g,按“2.1.3”項制得6份供試品溶液,用0.45 μm微孔濾膜過濾,取其濾液20 μL進樣分析。測定結果顯示,樣品中化合物 Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin含量的RSD(n=6)分別為0.92%、1.17%、1.25%、1.62%,說明重復性良好。
2.1.8 加樣回收率試驗 取尾葉香茶菜藥材均勻粉末6份,每份1.0 g,精密稱定,分別向其中加入對照品 Kamebakaurin 5.40 mg、excisanin A 2.48 mg、rabdokunmin C 1.18 mg、Kamebanin 1.18 mg,按“2.1.3”項制得6份加標提取液,各取25.0 mL稀釋至50 mL,得6份加標溶液。另取尾葉香茶菜藥材均勻粉末6份,每份1.0 g,同“2.1.3”項制得6份供試品溶液。取供試品溶液和加標溶液分別進樣分析,測定含量,計算回收率。結果化合物Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin的平均回收率分別為99.66%(RSD為0.89%)、99.75%(RSD為1.97%)、99.50%(RSD 為 2.27%)、99.86%(RSD為2.01%),表明方法的準確度符合要求(n=6)。
2.1.9 耐用性試驗 參照文獻[11]方法,分別考察流動相配比變化±2%、柱溫變化±2℃、檢測波長變化±2 nm、體積流量變化±10%,以及采用3根不同色譜柱(Diamonsil C18色譜柱,Shim-pack VP-ODS C18色譜柱,InertSustain C18色譜柱)進行測定時,儀器色譜行為的變化。結果顯示,不同條件下所測得四種二萜類成分含量的RSD均在2.3%以內,分離效果良好。因此,尾葉香茶菜中的四種二萜類成分測定條件較寬,具有較好的耐用性[12]。
2.2 相對校正因子(fk/s)的計算
2.2.1 多點校正法 參照文獻[11-13]方法,按照公式fk/s=(Cs×Ak)/(Ck×As)計算,得到其他成分相對于Kamebakaurin的fk/s。結果見表2。同時可根據公式Ck′=(Cs×Ak′)/(fk/s×As)計算其他三種待測成分的質量濃度。
2.2.2 斜率校正法 參照文獻方法[11],按照斜率校正法公式fk/s=ak/as計算(其中as為“2.1.4”項下所得標準曲線內參物斜率,ak為“2.1.4”項下所得標準曲線其他對照組分斜率),得出excisanin A、rabdokunmin C、Kamebanin相對于Kamebakaurin的 fk/s分別為0.952、0.933、1.042。同時可根據公式Ck′=Ak′/(as×fk/s)計算其他三種待測成分的質量濃度。
2.3 fk/s重現性考察
實驗考察了兩種儀器下3根色譜柱(Diamonsil C18色譜柱,Shim-pack VP-ODS C18色譜柱,InertSustain C18色譜柱)對fk/s的影響。結果見表3。
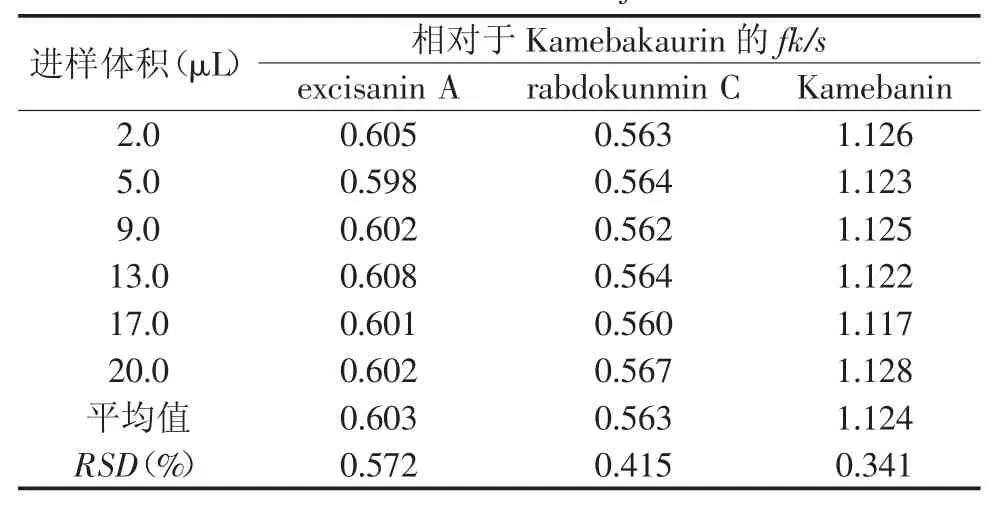
表2 以Kamebakaurin為參照的fk/s(多點校正法)
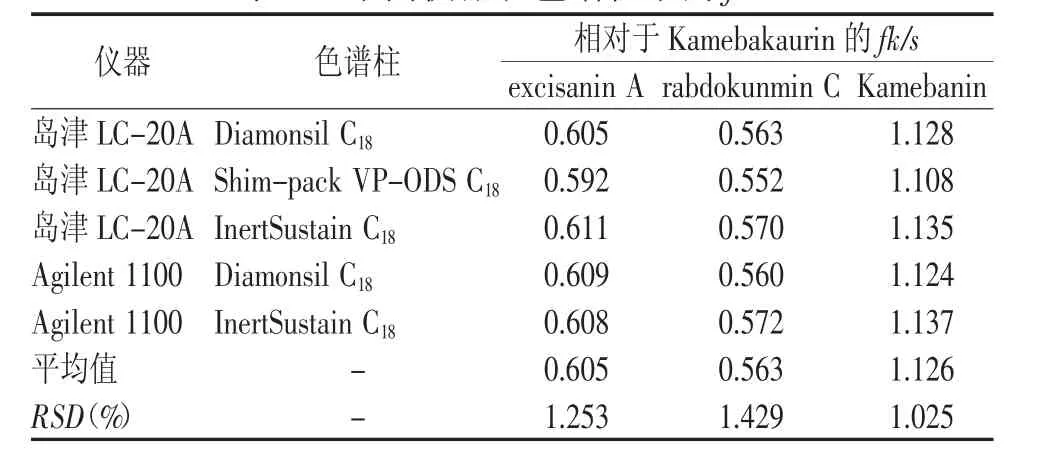
表3 不同儀器和色譜柱下的fk/s
2.4 待測組分的色譜峰定位
利用相對保留時間進行定位,即以Kamebakaurin為基準峰,計算不同儀器和色譜柱下excisanin A、rabdokunmin C、Kamebanin峰相對保留時間。結果見表4。
2.5 一測多評法與外標法測定結果的比較
分別精密吸取8批尾葉香茶菜供試品溶液各20 μL,進樣分析,記錄 Kamebakaurin、excisanin A、rabdokunmin C及Kamebanin的峰面積,用外標法和兩種一測多評法分別計算四種組分的含量,結果見表5。可得,尾葉香茶菜中的4種二萜類成分在不同采收期、不同采收地含量差異較大。采用t檢驗法,對多點校正法與外標法得到的尾葉香茶菜藥材中 excisanin A、rabdokunmin C、Kamebanin的含量進行比較,P>0.05,表明兩種方法測得的含量差異無統計學意義;多點校正法與外標法所得含量間的相對標準偏差均小于5%,說明多點校正法應用于尾葉香茶菜藥材的多指標成分質量評價中是可行的。采用t檢驗法,對斜率校正法與外標法得到的尾葉香茶菜藥材中以上3種二萜類成分的含量進行比較差異有統計學意義(P<0.05),表明兩種方法測得的含量差異有統計學意義;多點校正法與外標法所得含量間的相對標準偏差部分大于5%,說明斜率校正法應用于尾葉香茶菜藥材的質量評價有欠妥當。
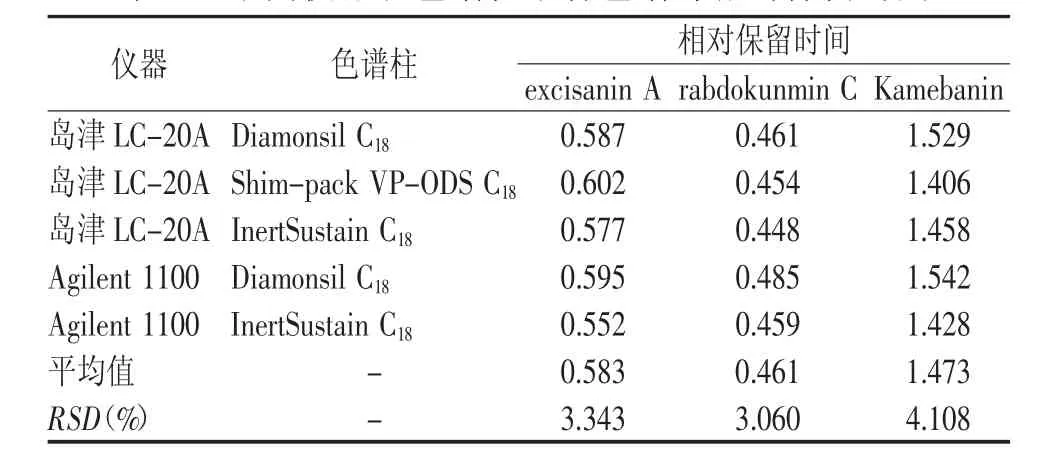
表4 不同儀器和色譜柱下各色譜峰相對保留時間
3 討 論
考察了水、乙醇、甲醇3種提取溶劑,室溫冷浸、超聲提取、回流提取、索氏提取、微波提取5種提取方法,結果以甲醇加熱回流提取的效率最佳,樣品色譜圖譜特征峰最為明顯,因此本實驗選用甲醇加熱回流提取。
Kamebakaurin、excisanin A、rabdokunmin C 及Kamebanin是尾葉香茶菜的主要特征性成分,在藥材中含量較高,藥理活性顯著,是評價尾葉香茶菜藥材質量的適宜指標。因此實驗中選取這4種化合物作為質控指標,并建立它們之間的相對校正因子fk/s。鑒于化合物Kamebakaurin在尾葉香茶菜中含量最高、易于制取、藥效顯著,實驗中選用Kamebakaurin為內參物,建立該成分與其他3種活性成分間的相對校正因子fk/s。
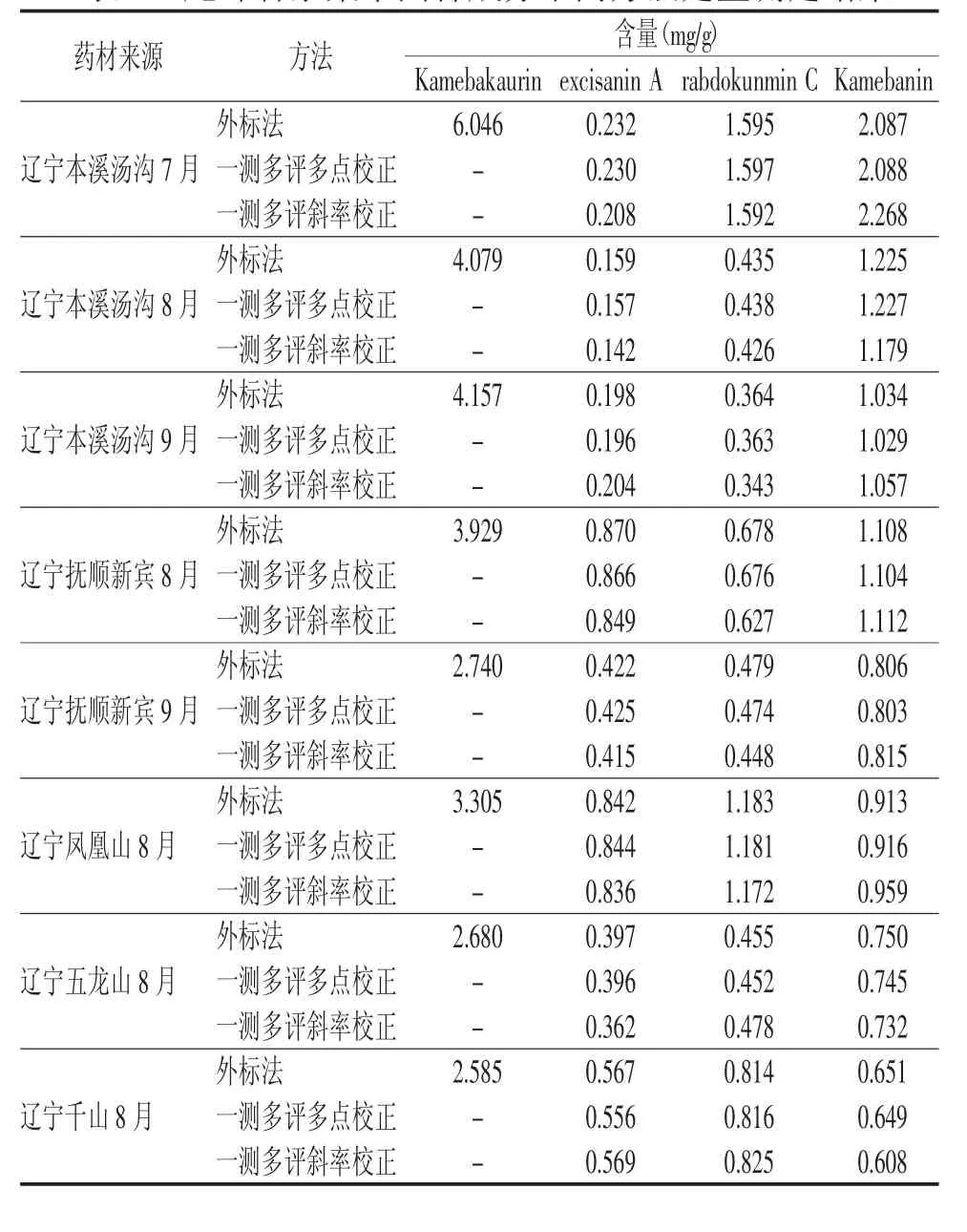
表5 尾葉香茶菜中四種成分不同方法定量測定結果
本實驗首次采用兩種一測多評法多點校正法和斜率校正法對尾葉香茶菜藥材的4種二萜類成分同時進行含量測定。斜率校正法作為其中一種一測多評法進行待測組分含量計算時,由于不同儀器峰面積計算方式不一,使得定量因子fk/s出現一定差異,所以每臺儀器的fk/s需單獨建立,在評價中存在一定的局限性。從表5可得,用斜率校正法所計算的結果與外標法所得結果差異有統計學意義(P<0.05)。而多點校正法所計算的結果與外標法所得結果差異無統計學意義(P>0.05),且不同實驗條件下各成分間的fk/s重現性良好(RSD<5%),說明多點校正法在對照品缺乏的前提下,可以作為一種方便、快捷、準確的方法應用于尾葉香茶菜藥材多成分的含量測定和藥材質量的初步評價。
實驗中利用相對保留時間進行待測組分色譜峰定位(RSD<5%),但鑒于尾葉香茶菜藥材色譜峰較復雜,為了保證QAMS色譜峰定位的準確性,應當結合光譜法或質譜法進行定位。
