LC-MS/MS法測定煙草中7種高級脂肪酸
2018-03-18 05:17:20馬麗伊徐志強田振峰趙愛俠淦五二
中國煙草學報 2018年6期
馬麗伊,徐志強,田振峰,趙愛俠,淦五二*
1 中國科學技術大學化學系,合肥 230026;
2 安徽中煙工業有限責任公司技術中心,合肥 230088
高級脂肪酸對煙葉品質具有重要影響[1-6],并且是評價烤煙品質的有效指標[6]。煙草中飽和高級脂肪酸能增加煙氣的脂肪或蠟味,其中,肉豆蔻酸能柔和煙氣,與煙香諧調,提高甜而醇和的風味;棕櫚酸有甜的蠟脂氣息,能增加豐滿度;但亞油酸、亞麻酸等不飽和脂肪酸會增加煙氣刺激性[7]。
煙草及煙草制品中高級脂肪酸的測定方法主要有氣相色譜法[8-11]和液相色譜法[12-13]。采用氣相色譜分析時,衍生化樣品操作較麻煩,處理步驟多,而且會對樣品造成損失。對于液相色譜法來說,由于脂肪酸紫外吸收很弱,用紫外檢測器檢測時需柱前衍生化,但在衍生過程中易發生異構化、氧化、分解等副反應且很難保證衍生化完全。用蒸發光散射檢測器可直接檢測脂肪酸,但靈敏度低,無法檢測到低含量的高級脂肪酸。
液相色譜-串聯質譜法(LC-MS/MS)檢測靈敏度高、選擇性好,是非揮發性成分定性、定量測定的有效手段[14-15]。本文建立了一種快速、準確的LC-MS/MS測定高級脂肪酸檢測方法。所研方法樣品前處理相對簡單,無需衍生化,提取的樣品可直接進樣分析,適用于分離、定量煙草中高級脂肪酸。
1 材料和方法
1.1 材料和儀器
(1)試劑:十七烷酸(98%)、亞麻酸(99%,上海阿拉丁生化科技股份有限公司),油酸、棕櫚酸、肉豆蔻酸(分析純,國藥集團有限公司),硬脂酸(98%,英國Alfa Aesar 公司),月桂酸、亞油酸(99%,美國Acros Organics 公司),氫氧化鉀、鹽酸(分析純,國藥集團有限公司),其余所用試劑均為色譜純。煙葉樣品: 不同等級(C2F,B2F,X2F,X3F,C3F)烤煙樣品來自云南梁河。
(2)儀器:Symbiosis Pico高效液相色譜儀(荷蘭Spark公司),4000 Q Trap質譜儀(美國AB公司),Milli-Q超純水儀(美國Millipore公司),AL204-IC型電子分析天平(0.1 mg,瑞士Mettler Toledo公司)。
1.2 標準溶液的配制
分別準確稱取7種高級脂肪酸標準品各10 mg,用甲醇溶解定容至50 mL,制得0.20 mg/mL單一標準儲備液。用甲醇準確配制混合標準儲備液(月桂酸:1.0 μg/mL;肉豆蔻酸:2.0 μg/mL;棕櫚酸:50 μg/mL;硬脂酸:10 μg/mL;亞麻酸:60 μg/mL;亞油酸:42 μg/mL;油酸:20 μg/mL)儲存于棕色玻璃瓶中。
準確配制十七烷酸(0.20 mg/mL)內標標準儲備液,取一定量的內標儲備液,用甲醇稀釋定容,制得內標工作液(10 μg/mL)。
1.3 樣品處理與分析
稱取樣品0.5 g于100 mL 圓底燒瓶中,加入40 mL含3 mol/L KOH的甲醇溶液,于60℃水浴中回流皂化60 min,冷卻至室溫。過濾溶液至錐形瓶中,加入20 mL水,混勻,用36%(質量分數)鹽酸調節濾液pH值至3.0 ~ 4.0。用含內標的二氯甲烷溶液萃取3次,每次15 mL,合并二氯甲烷層,以30 mL超純水反萃取有機相,將有機相用氮氣吹干,殘余物用甲醇溶解并定容至50 mL,移取0.2 mL上述甲醇溶液稀釋至10 mL,經0.22 μm有機相濾膜過濾后進行LC-MS/MS分析。
1.4 儀器條件
色譜條件:Hypersil Gold C18色譜柱(2.1 mm×150 mm,3 μm),流動相 A:5%(ν/ν)緩沖溶液的水溶液;流動相 B:5%(ν/ν)緩沖溶液、55%(ν/ν)乙腈、40%(ν/ν)異丙醇。緩沖溶液配制:用濃氨水調節100 mM冰醋酸至pH=5。運行時間30 min,柱溫20℃,進樣體積 5 μL。梯度洗脫條件:0 ~ 20 min,70%B~100% B;保持 5 min;25~30 min,100% B~70% B,流速為0.2 mL/min。
質譜條件:電離方式為電噴霧負離子模式(ESI-);離子源溫度:550℃;電噴霧電壓:-4000 V;氣簾氣:10 psi;掃描時間:0.1 s;離子源輔助氣1:50 psi;離子源輔助氣2:50 psi;檢測方式:“準分子”多反應監測過渡(即采用相同的離子作為母離子和子離子)。
2 結果與討論
2.1 儀器條件的建立
LC-MS/MS定量測定高級脂肪酸常用檢測模式有MRM[15](多反應監測)和SIM[16-17](選擇離子掃描)模式。MRM模式分析高級脂肪酸雖然可以減少煙草中基質干擾,但分析物在負離子模式下,加碰撞能產生的碎片離子靈敏度很低,無法滿足定量條件的要求。SIM模式比MRM模式在高級脂肪酸分析中有更好的靈敏度,但對于煙草樣品會有更多的分析干擾。“準分子”MRM過渡模式是采用相同的離子作為母離子和子離子,既解決了分析干擾因素又提高了分析靈敏度。本文比較了以上3種檢測模式,結果如表1所示。從表1可知,“準分子”MRM過渡模式響應值高于MRM模式,與SIM模式相比,大部分目標物響應值略高。因此,本文采用“準分子”MRM過渡模式。
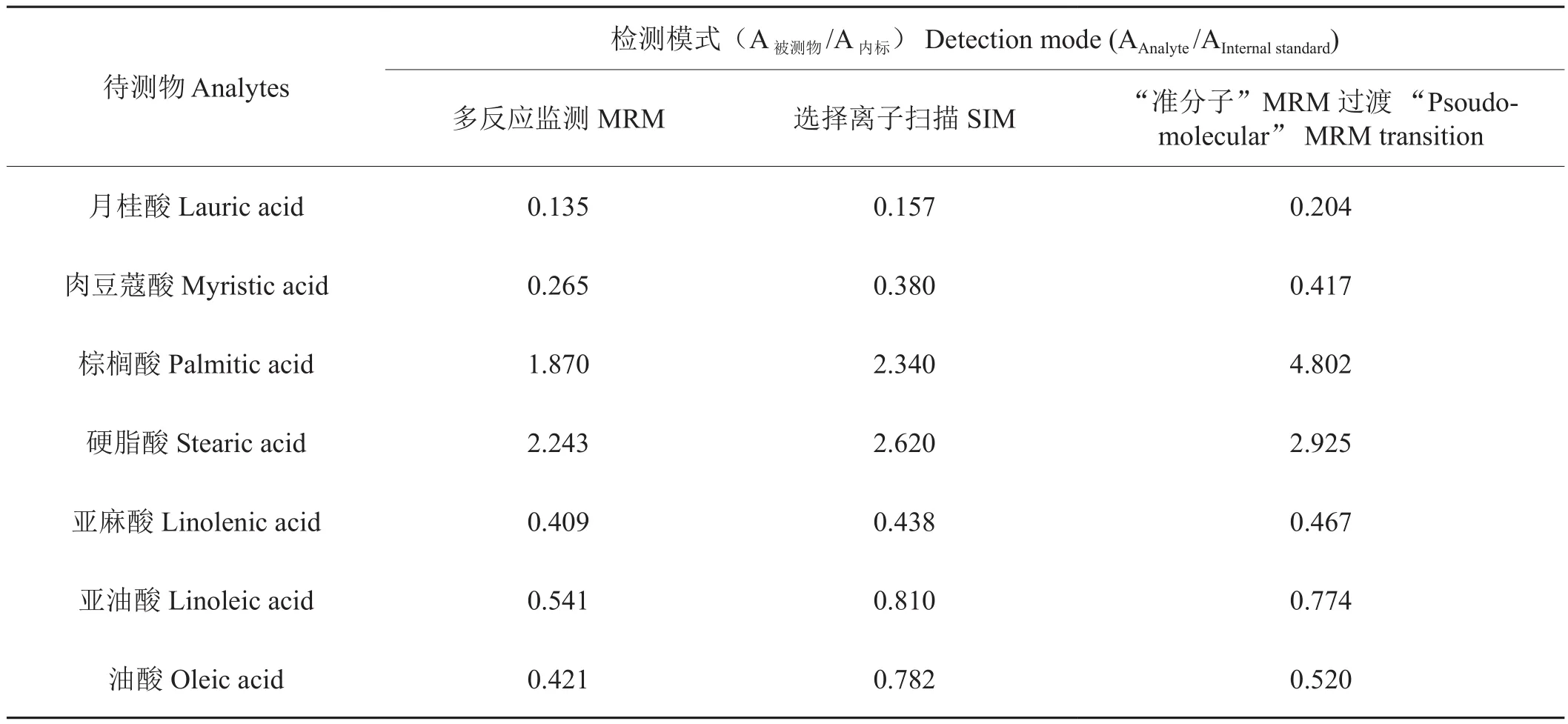
表1 “準分子”MRM過渡、SIM和MRM模式對比Tab.1 Comparison of the MRM, SIM and “Pseudo-molecular” MRM transition mode
2.2 離子源優化
本文分別采用ESI源和APCI源進行離子源優化。由圖1可以看出,除棕櫚酸外,其它被測物的ESI源信號強度均高于APCI源,因此本文采用ESI源。
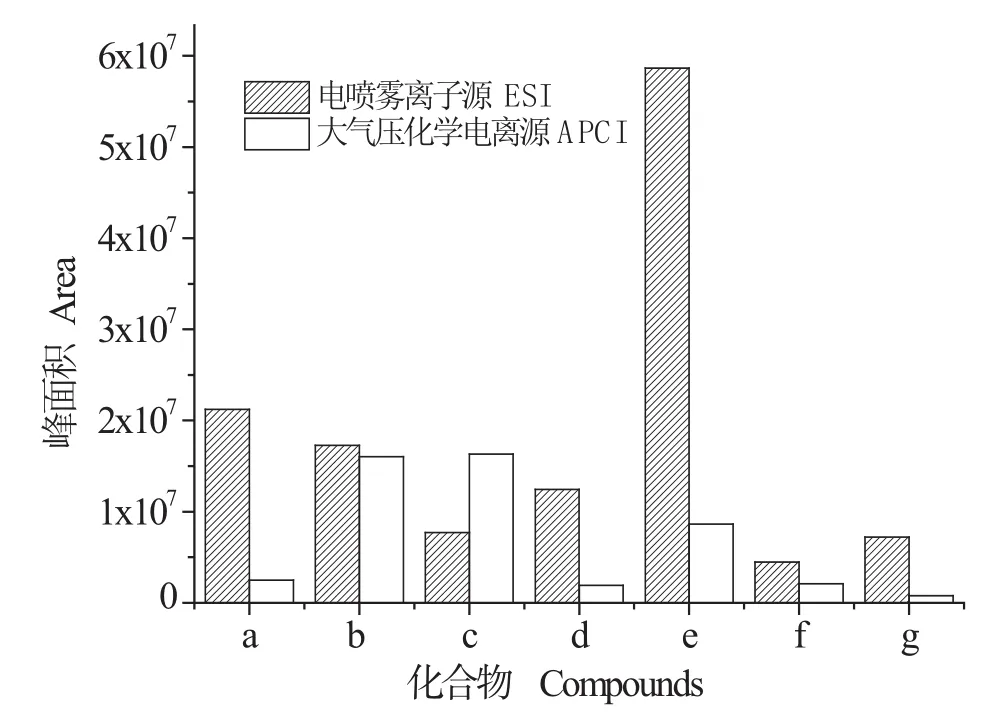
圖1 對比使用ESI和APCI接口目標化合物的靈敏度Fig. 1 Sensitivity of the target compounds using the ESI and APCI interfaces
采用負離子模式對標準樣品進行一級質譜分析,從而確定母離子。然后對各質譜條件進行優化,以提高靈敏度使得信號穩定。最終確定質譜最佳檢測條件,各化合物的優化結果如表2所示。
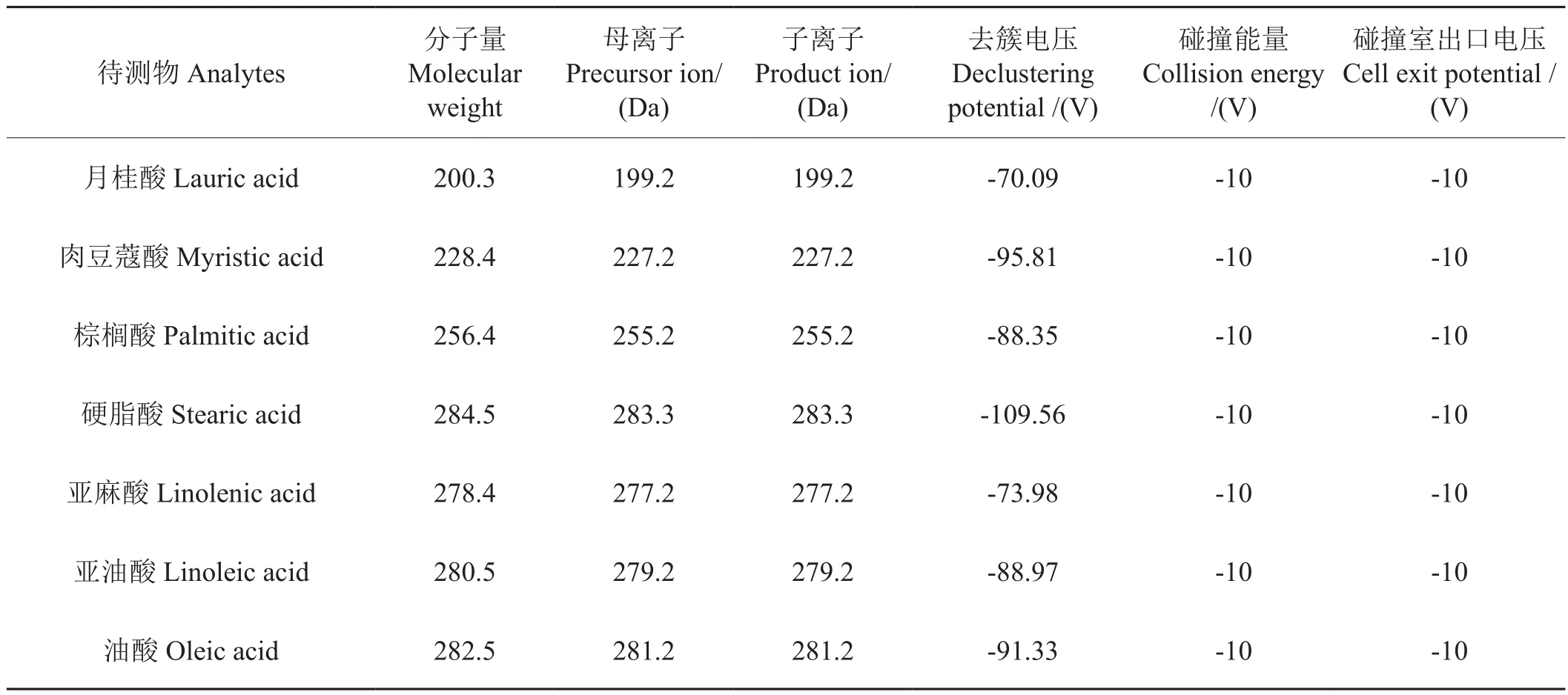
表2 質譜優化條件Tab.2 Investigated compounds with their specific MS/MS parameters
2.3 流動相的選擇
實驗考察了甲醇-水,乙腈-水,緩沖溶液的水溶液-乙腈/異丙醇/緩沖液體系作流動相對分離效果的影響。結果如圖2所示,乙腈-水作為流動相時,峰型較差,響應較低;甲醇-水作流動相時的峰型較好,響應較高,干擾峰較少,但目標物不能有效分離。改用A相為5%緩沖溶液的水溶液,B相為5%緩沖溶液、55%乙腈、45%異丙醇作流動相。加入緩沖鹽溶液調節溶液pH值高于pKa使得脂肪酸易脫質子產生負離子,加入異丙醇后調節有機相極性,實現了各目標物完全分離,且具有較好的方法靈敏度。因此選用此條件作為流動相分析條件。
2.4 前處理過程的優化
根據文獻報道[13],選擇甲醇作皂化溶劑,分別對氫氧化鉀濃度,皂化溫度,反應時間,萃取劑提取效率和萃取劑體積進行優化。考察了皂化溫度分別為40,50,60和70℃時對各化合物測定值的影響。結果表明,當皂化溫度從40℃增加到60℃時,各物質的測定值顯著增加,繼續升高溫度,各物質含量變化趨于平緩。因此選擇皂化溫度為60℃。分別比較了反應時間為20,40,60和80 min時的各化合物測定值。結果表明,隨著反應時間的增加,各化合物測定值逐漸增加。當反應時間從60 min增加到80 min時,各物質測定值變化不大。表明反應時間為60 min時皂化反應進行完全。因此選擇反應時間為60 min。
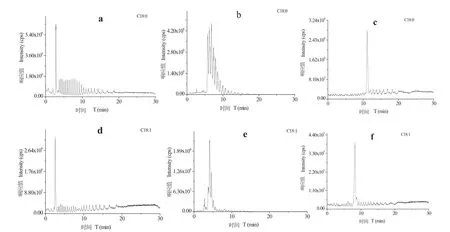
圖2 三種流動相對比圖。a b c分別為甲醇-水,乙腈-水,緩沖溶液的水溶液-緩沖溶液/乙腈/異丙醇作流動相,a b c為硬脂酸色譜圖,d e f為油酸色譜圖Fig. 2 Comparison of LC-MS/MS chromatograms of three mobile phases a MeOH-H2O, b MeCN-H2O, and c buffer/H2O(5/95)-buffer/MeCN/IPA(5/55/40); a b c Stearic acid chromatogram, d e f Oleic acid chromatogram
2.4.1 氫氧化鉀濃度的選擇
分別用1、2、3、4 mol/L KOH的甲醇溶液進行皂化實驗(表3)。結果顯示KOH的甲醇溶液濃度由1 mol/L上升到3 mol/L 時,高級脂肪酸的測定值顯著增大,隨后趨于平緩。因此本文選用3 mol/L KOH的甲醇溶液作皂化溶劑。
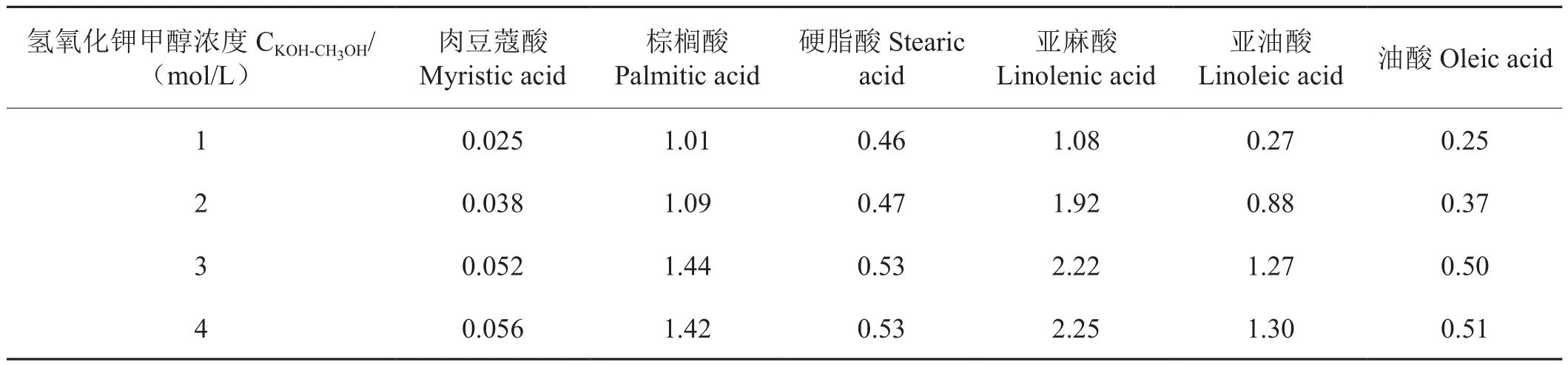
表3 不同濃度KOH的甲醇溶液下各高級脂肪酸的測定值Tab.3 Effect of KOH-CH3OH concentration on high fatty acids mg/g
2.4.2 萃取溶劑的選擇
本實驗選取了環己烷、無水乙醚、二氯甲烷、乙酸乙酯、正己烷作為萃取液優化高級脂肪酸萃取條件,發現無水乙醚和乙酸乙酯不能與水相較好地分層,因此比較了環己烷、二氯甲烷、正己烷對目標物萃取效率的影響。為防止環己烷、二氯甲烷、正己烷溶劑和流動相不匹配,將萃取液用氮氣吹干后用甲醇定容,在相同條件下進行分析,結果見圖3。對于肉豆蔻酸和亞油酸,環己烷萃取效率略高,但其他化合物二氯甲烷萃取效率最高,因此最終選用二氯甲烷為萃取溶劑。
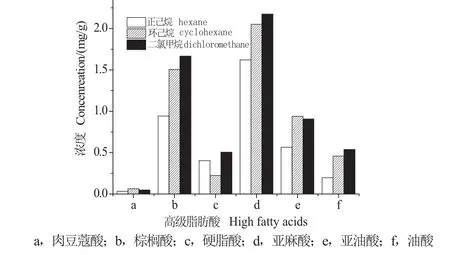
圖3 不同提取試劑對各高級脂肪酸的影響Fig.3 Effect of extraction reagents on high fatty acids
2.4.3 萃取溶劑體積選擇
固定樣品量為0.5 g,分別比較了萃取溶劑體積為10、12、15、18 mL時的萃取效率(表4)。結果表明,隨著萃取溶劑增加,提取出的被測物含量逐漸增加。當萃取溶劑達到15 mL時萃取基本完全。因此使用萃取溶劑體積為15 mL。
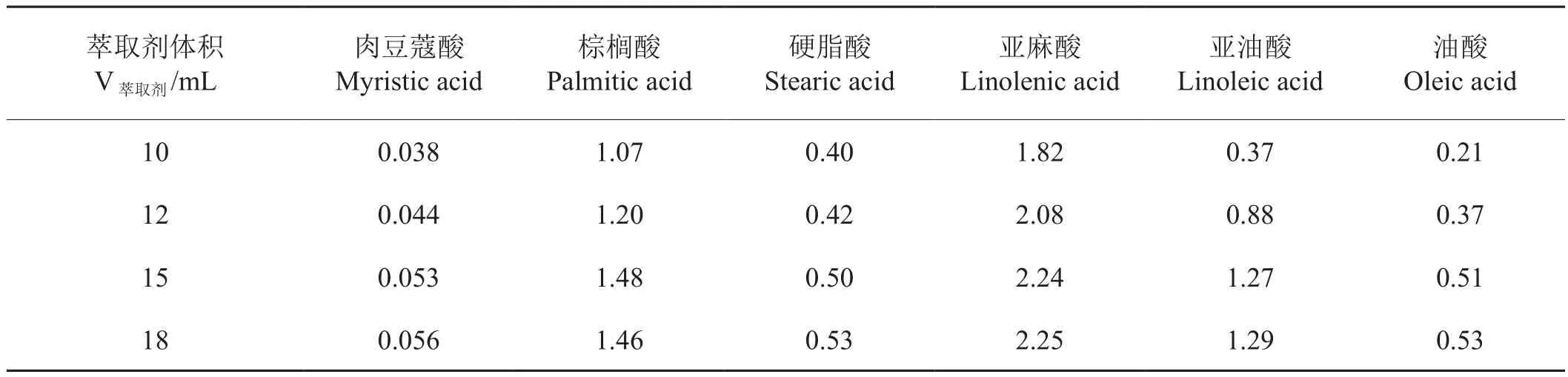
表4 不同體積萃取溶劑下各高級脂肪酸的測定值Tab.4 Effect of Extraction Solvents Volume on high fatty acids mg/g
2.5 方法的標準曲線、精密度及回收率
準確配制系列混合標準溶液,按照1.3所述條件進行LC-MS/MS分析,混合標準溶液選擇離子流圖如圖4所示。內標法線性回歸得到標準曲線方程、相關系數、回收率、LOQ(表5)。各目標化合物具有良好的相關性,適合定量分析。將最小濃度的標準溶液重復進樣10次得到標準偏差,以3倍標準偏差為方法的檢測限,10倍標準偏差為定量限。各物質的LOD范圍為0.0004~0.007 mg/g,LOQ范圍為0.001~0.02 mg/g。在空白樣品中添加7種高級脂肪酸混合標樣,添加濃度0.02~1.2 mg/g之間,重復進行5次實驗,得出平均回收率為87.5%~109%,相對標準偏差在 1.1%~4.7%之間(表5)。
圖4 混合標樣和內標選擇離子流色譜圖Fig.4 Chromatogram of mixed standard and internal standard
2.6 本方法與氣質方法比較
分別采用GC-MS[11]和LC-MS/MS法測定高級脂肪酸(表6)。結果表明,在線性范圍內,相關系數均大于0.98,說明兩種方法均具有良好的線性關系。LC-MS/MS靈敏度優于GC-MS,但回收率和精密度略差于GC-MS,兩種方法均滿足定量分析要求。高級脂肪酸屬于低揮發性物質,因此不適合直接進行GC-MS分析,需進行衍生化,反應時間2 h,衍生化副產物較多,有時可干擾目標物的檢出,且無法分別定量亞麻酸和油酸。而LC-MS/MS法皂化后直接測定,無需衍生化,減少樣品用量,分離時間快且有效縮短檢測時間。
對于高級脂肪酸含量測定,本方法是通過皂化使煙草中酯結合態脂肪酸轉化為游離態脂肪酸,經提取得到煙草中總的脂肪酸含量;而GC-MS方法是通過酯化使游離態脂肪酸轉化為酯結合態脂肪酸,經提取后得到總的脂肪酸含量。比較上述兩種方法對煙草樣品進行分析(表7)。由表7可知,兩種方法測定樣品均未檢出月桂酸,不同烤煙中高級脂肪酸含量存在一定差異。本文方法測定樣品中高級脂肪酸含量分別為:肉豆蔻酸(0.018~0.073 mg/g)、棕櫚酸(1.43~1. 87 mg/g)、硬脂酸(0. 24~0. 55 mg/g)、亞麻酸(2.17~2.99 mg/g)、亞油酸(0.56~1.28 mg/g)、油酸(0. 43~0. 86 mg/g)。氣質測定結果為肉豆蔻酸(0.019~0.055 mg/g)、棕櫚酸(1.63~2.19 mg/g)、硬脂酸(0.37~0.46 mg/g)、亞油酸(1.12~1.64 mg/g)、亞麻酸+油酸(2.55~3.78 mg/g)。總體看來,對于肉豆蔻酸,液質方法測定含量高于氣質方法,其他化合物氣質測定含量高于液質。
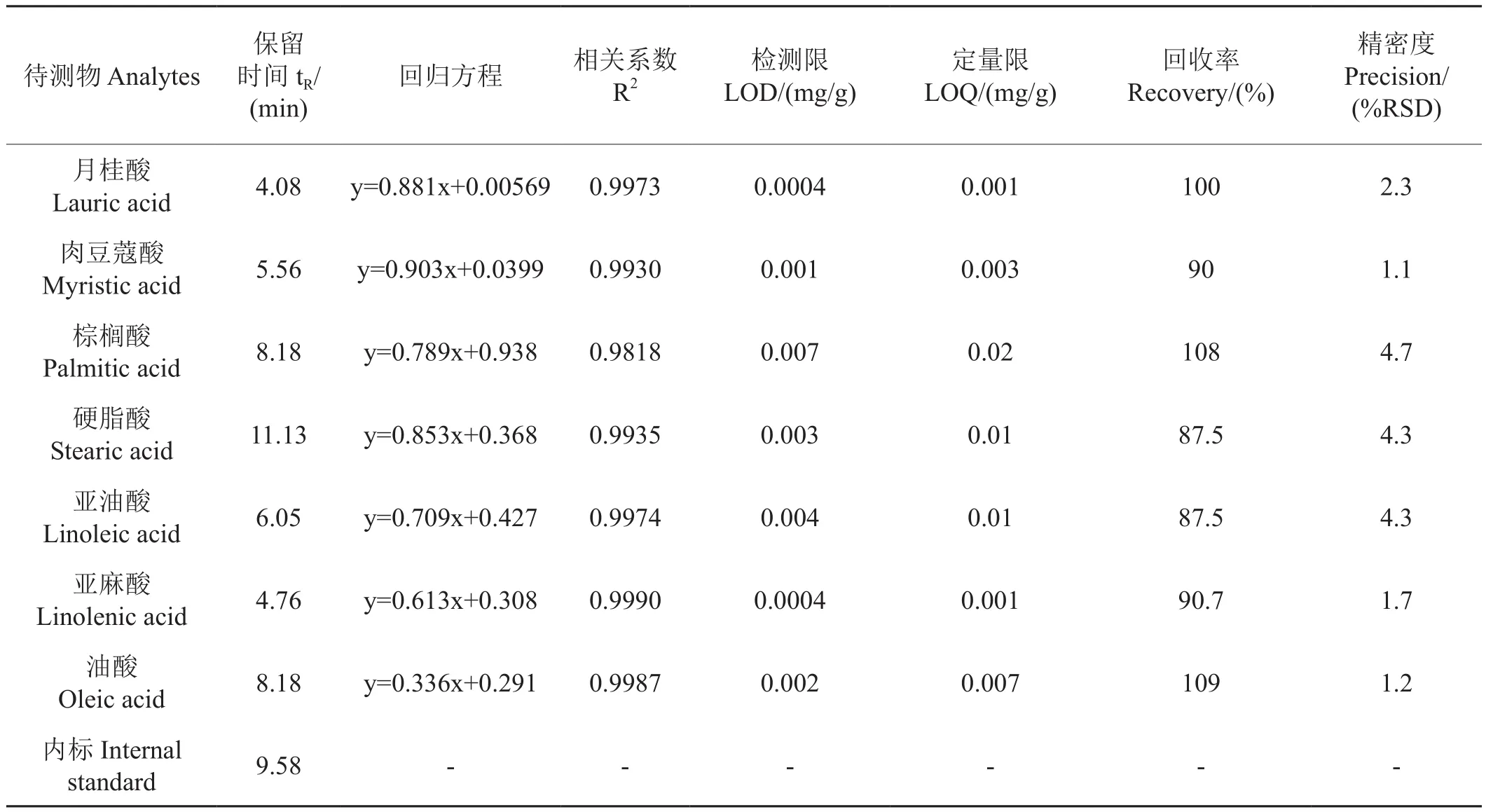
表5 7種高級脂肪酸的相關參數Tab.5 Quality parameters of 7 high fatty acids
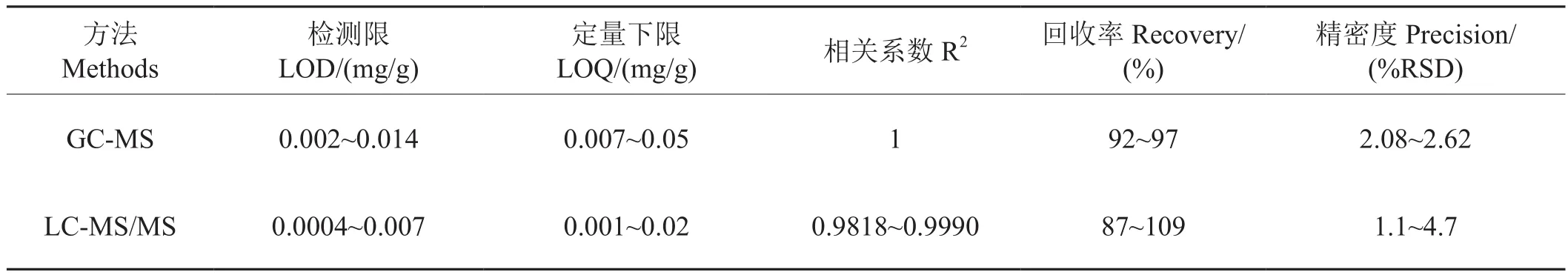
表6 比較2種高級脂肪酸的測定方法Tab.6 Comparison of two methods for the determination of high fatty acids
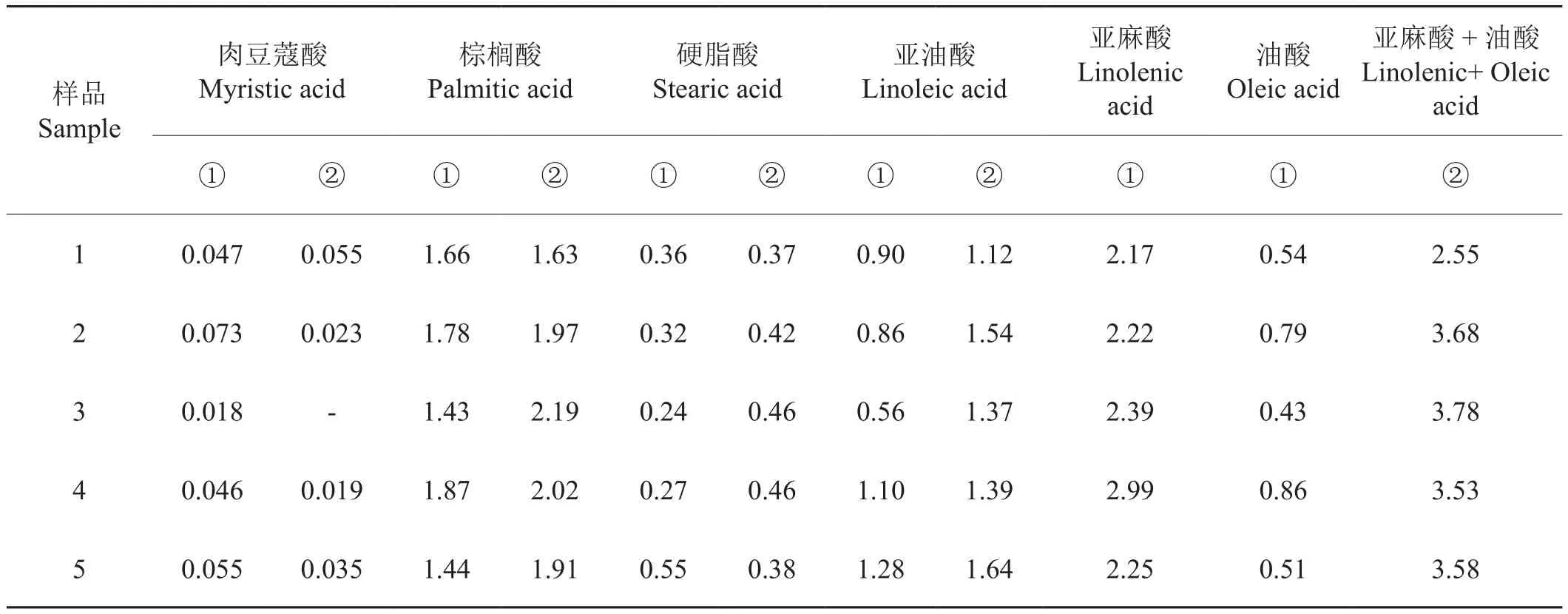
表7 比較2種方法測定煙草樣品中高級脂肪酸含量Tab.7 Comparison of two methods for the determination of high fatty acids in tobacco mg/g
3 結論
建立了一種高效液相色譜-串聯質譜內標法測定煙草中高級脂肪酸含量的方法。該法快速、準確,具有精密度高、準確度好、操作簡單等特點,避免了衍生化處理對樣品造成損失的問題,滿足煙草中高級脂肪酸定量分析。
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
兒童故事畫報(2019年5期)2019-05-26 14:26:14
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
Coco薇(2016年2期)2016-03-22 02:42:52
當代化工研究(2016年9期)2016-03-20 16:22:08
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
聲屏世界(2014年6期)2014-02-28 15:18:09
中國煙草學報(2012年5期)2012-04-12 06:21:18
