miR-145反向調節WT-p53對前列腺癌細胞EMT和干細胞特性的抑制作用
2018-05-22 07:30:58江展慶
安徽醫科大學學報 2018年4期
陳 松,郭 瑋,江展慶,劉 琦
骨轉移是許多惡性腫瘤的終末期并發癥,其中多達65%~75%的晚期前列腺癌患者罹患骨轉移,且確診骨轉移后的中位生存時間僅為12~53個月[1]。調控腫瘤細胞轉移的基因一直被廣為研究,其中p53已被證實能夠促進細胞凋亡、衰老、調節細胞周期。野生型p53(wild-type p53, WT-p53)因突變而導致功能丟失,影響了多種癌癥的發生和發展[2]。p53能夠通過調節癌細胞的上皮-間充質轉化(epithelial-mesenchymal transition, EMT)和腫瘤干細胞特性抑制其遷移和侵襲[3]。EMT是腫瘤細胞發生轉移的關鍵步驟之一,而WT-p53正是通過調節E-cadherin、Snail、Slug、ZEB1/2等EMT相關的轉錄因子來發揮作用[4]。腫瘤干細胞是整個腫瘤中具有自我更新能力、可以永生化增殖和成癌的一類異質細胞系,這些功能被定義為“干細胞特性”(簡稱干性)[5]。越來越多的證據[3,6]表明,癌細胞的干細胞特性與腫瘤的轉移密切相關,而p53則可通過調節相關基因的表達來調節癌細胞的干細胞特性。該前期研究[7-8]已經證明miR-145能夠通過抑制癌細胞的EMT和干性來抑制前列腺癌的骨轉移。現已知miR-145由WT-p53直接調控[9],由此推斷:WT-p53基因可能在前列腺癌細胞的EMT和干性轉化過程中發揮了重要的調節作用,而miR-145則中介了WT-p53的這一功能。該文針對該假說進行驗證,最終證明WT-p53抑制了PC-3細胞的EMT和干性,miR-145則反向調節了WT-p53對前列腺癌細胞EMT和干性的抑制作用。
1 材料與方法
1.1細胞培養骨轉移性前列腺癌細胞系PC-3(美國典型培養物保藏中心);在含有10%胎牛血清的F-12培養基中培養和擴增(美國HyClone公司)。細胞在孵育溫度37 ℃并含5% CO2的培養箱中培養。
1.1.1質粒和瞬時轉染 WT-p53和抗-miR-145的表達質粒(廣州銳博生物有限公司)。轉染前將2×105個細胞接種到6孔板中無血清培養24 h。使用Lipofectamine 2000(美國Invitrogen公司)進行轉染。轉染4~6 h后去除轉染培養基,將細胞置于含10%胎牛血清的培養基中培養48 h后進行下一步實驗。
1.1.2實時熒光定量PCR(real-time PCR, RT-PCR) 該方法按照ALL-IN-ONETM的miRNA定量RT-PCR檢測試劑盒的說明書進行。總RNA使用RNeasy試劑盒(美國Qiagen公司)從細胞中提取。通過添加多聚A序列逆轉錄所有樣本,并用特定的引物對WT-p53和HSA-miR-145進行RT-PCR分析。每個樣品設3個復孔,U6設為內參。重復3次并用Schmittgen和Livak的2-ΔΔCT方法計算目的基因的相對表達量[10]。
1.2Westernblot分析將細胞接種在6孔板中培養,待24~48 h后細胞融合至60%~70%時,在各培養孔中加入樣品緩沖液[62.5 mmol/L的Tris-HCl(pH6.8),2% SDS,10%甘油,5%β-巰基乙醇]。各泳道中加入等量樣品蛋白并通過SDS-聚丙烯酰胺電泳分離。將分離過的蛋白質轉移到PVDF膜(美國Millipore公司)后,5%脫脂奶粉室溫下封閉1 h,并與一級抗體(1 ∶1 000)4 ℃過夜進行結合。膜在TBS-T緩沖液洗滌3次(每次10 min),并在室溫下與二級抗體結合40 min,再次TBS-T洗滌3次后ECL系統化學發光、顯影。
1.3劃痕實驗PC-3細胞均勻接種到6孔板中使之在24 h內達到完全融合。無血清饑餓24 h后,用無菌100 μl吸頭的尖端進行劃痕,PBS洗滌2次,并在含10%胎牛血清的培養基中培養。分別在0、24 h的時間點拍攝PC-3細胞遷移及劃痕愈合的圖像(×40)。
1.4侵襲功能檢測侵襲功能使用涂有基質膠(Matrigel,美國BD Biosciences公司)的8 mm孔徑Transwell小室(美國Corning公司)系統進行檢測。將細胞制備成單細胞懸液并置于無血清培養基中,將1.5×105個細胞加入到上室中,下室填充含10%胎牛血清的培養基。培養箱孵育24~48 h后,將穿過多孔膜的細胞用4%多聚甲醛固定并蘇木精染色,顯微鏡(×100)下計數細胞。
1.5黏附實驗將50 μl纖連蛋白(50 μg/ml)加入96孔板各孔,培養箱中加熱1h以包被,并用1%胎牛血清白蛋白封閉。將懸浮細胞以1.5×104個/孔的密度接種到各孔中。溫育30 min后洗除非貼壁細胞,將貼壁細胞用4%多聚甲醛固定,蘇木精染色及顯微鏡(×100)下計數。
1.6成球實驗將各組細胞(500個/孔)接種到6孔板(美國Corning公司)中,在無血清的DMEM-F12(美國Bio Whittaker, Lonza公司)中懸浮培養,培養液中補充B27(1 ∶50,美國Invitrogen公司),EGF(20 ng/ml,美國BD Biosciences公司),胎牛血清白蛋白(0.4%)、胰島素(4 mg/ml)(美國Sigma公司)。培養10~14 d后,于顯微鏡下計數PC-3細胞球(緊密、球形、非粘附狀態且直徑>50 μm)的數量。細胞球形成率(%)=集落數/接種細胞數×100%。
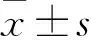
2 結果
2.1WT-p53能夠抑制PC-3細胞的侵襲能力本研究將高表達WT-p53的質粒轉染進入PC-3細胞中,并通過RT-PCR和Western blot分別驗證轉染成功(圖1,F=117.8,P<0.01),以備進一步實驗。在功能學實驗中,本研究顯示WT-p53高表達的PC-3 細胞,其侵襲能力較對照組下降了45.7%(圖2,F=40.18,P<0.05),而黏附能力卻增加了2.87倍(圖3,P<0.01)。同時,WT-p53高表達的PC-3細胞使劃痕愈合的速度也顯著低于對照組(圖4,F=379.08,P<0.05)。
本研究上調PC-3中WT-p53的表達量后,再轉染miR-145的反義抑制序列(抗-miR-145)。結果表明:抗-miR-145不僅能夠下調WT-p53的表達(圖1,F=117.8,P<0.01),還能阻斷其功能,表現為PC-3的遷移速度和侵襲能力明顯增加,黏附能力顯著降低(圖2~4,F=40.18,P<0.05)。

圖1 p53高表達和miR-145低表達對PC-3細胞中p53表達水平的影響
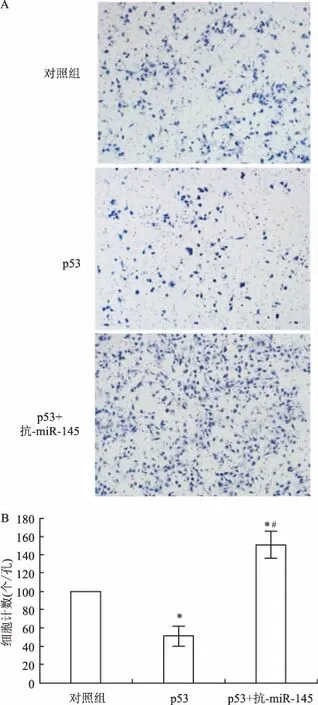
圖2 p53高表達和miR-145對PC-3細胞侵襲相關細胞生物學行為的影響 亞甲蘭染色×100
2.2WT-p53抑制了PC-3的EMT而抗-miR-145可逆轉此作用E-Cadherin作為上皮細胞的標記蛋白之一,往往在細胞發生EMT時表達減少。但本實驗中WT-p53在PC-3細胞中高表達時,E-cadherin的表達量也相應增加。而Fibronectin和Vimentin等作為間充質細胞的標志物,在EMT過程中往往表達增加。而本實驗中WT-p53在PC-3細胞中的表達增加時,此二者的表達被顯著抑制(圖5)。此外,本研究還觀測到PC-3的細胞形態與EMT特性密切相關:隨著WT-p53基因的表達水平增高,PC-3細胞的形態從棒狀或長梭形的間充質細胞形態,逐漸轉化為鵝卵石狀或短紡錘形的上皮細胞形態(圖6)。

圖3 p53高表達和miR-145對PC-3細胞粘附能力的影響 亞甲蘭染色×40
2.3抑制miR-145的表達能夠阻斷PC-3細胞中WT-p53的作用如圖5所示,在WT-p53高表達的狀態下,抗-miR-145減少了上皮細胞標志物E-Cad-herin的表達,恢復了間充質細胞標志物Vimentin和Fibronectin的表達水平;同時也使得PC-3細胞由間充質細胞形態向上皮細胞形態轉換,見圖6。

圖4 p53高表達和miR-145低表達對PC-3細胞遷移能力的影響 ×40
2.4WT-p53抑制PC-3細胞成球而抗-miR-145可阻斷此效果各實驗組的PC-3細胞在非黏附條件下培養12 d之后,所有實驗組有癌細胞球形成,球體形成率分別是4.8%(對照組)和2.3%(PC-3/WT-p53),這說明WT-p53能夠抑制前列腺癌細胞形成非黏附球體(F=70.18,P<0.01),見圖7。更為重要的是,本研究還顯示抗-miR-145完全逆轉了WT-p53抑制成球的能力,使PC-3/WT-p53/抗-miR-145實驗組的成球率較PC-3/WT-p53實驗組提高了6.7%。
3 討論
已有充分的研究[7-8]證明了miR-145可以抑制前列腺癌的骨轉移,并參與調節EMT和前列腺癌細胞的干性,但其上游控制基因尚未有效闡明。而本研究顯示,不但WT-p53能夠抑制PC-3細胞的遷移、侵襲、EMT和腫瘤球形成,而且其上述抑制作用能夠由抗-miR-145阻斷并扭轉。由此得出結論:WT-p53能夠至少一部分通過調控miR-145以抑制腫瘤細胞的EMT和干性。

圖5 各組細胞表達EMT特征蛋白的差異
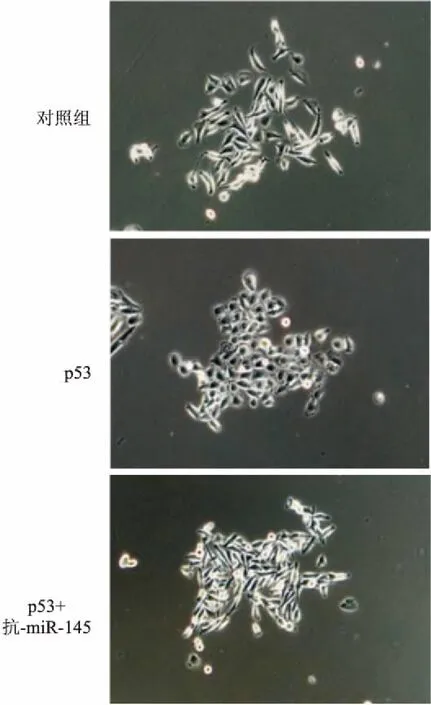
圖6 各組細胞形態差異 ×200

圖7 p53通過調控miR-145抑制PC-3細胞的腫瘤球形成能力 ×200
許多研究[11-12]已經證明,WT-p53基因除在調節細胞周期、細胞凋亡、衰老、DNA修復和自噬時發揮關鍵作用外,在調節腫瘤細胞轉移的機制中也起著至關重要的作用。有研究[11]顯示,p53和Rb發生聯合缺陷可能是使前列腺發生高侵襲度和高轉移度惡變的關鍵因素之一。此外,前列腺上皮細胞中PTEN/TP53的缺失也導致了其向多能祖細胞轉化和EMT化,惡性度增加[12]。這些發現均提示前列腺細胞中WT-p53的功能缺失可能通過提高細胞遷移能力、侵襲能力、EMT和干性以促進前列腺癌的發展和轉移。
更為重要的是,本研究還發現p53除上述途徑外,還可通過調節腫瘤細胞中microRNA的表達水平來發揮抑癌作用。已有類似的研究[13]證明,WT-p53的功能缺失導致miR-34a的表達量下降,而后者是Snail的直接靶向抑制因子,因此造成Snail依賴性的EMT在結腸癌、乳腺癌和肺癌細胞中被激活。這支持了本研究結論:不僅上調WT-p53能夠增強PC-3細胞中的miR-145表達水平,抗-miR-145也能夠逆轉由WT-p53抑制的PC-3細胞的EMT特性。這說明miR-145在WT-p53調控EMT和干性的過程中充當了中介作用。
已有研究[14]顯示WT-p53能夠抑制前列腺癌細胞中N-cadherin和ZEB2的表達,后兩者均是miR-145的目標靶基因,且均是EMT的轉錄因子。WT-p53對PC-3細胞中N-cadherin和ZEB2的抑制作用也均能夠被抗-miR-145逆轉。因此本研究推測,miR-145可能通過對N-cadherin和ZEB2的抑制來介導WT-p53對PC-3 細胞EMT的調節。
除了調控EMT之外,microRNA還可以作為p53的直接抑制靶基因以調節其抑制癌細胞干性的作用,例如p53可以通過調控miR-200c來調節KLF4和BMI1[3]。本研究之前的結果已經證明miR-145通過抑制CD44、Oct4、c-myc和Klf4以抑制PC-3細胞的干性[8],而本研究顯示,抗-miR-145還能夠逆轉WT-p53對PC-3細胞成球的抑制,這說明miR-145除了本身可以靶向抑制干性相關蛋白之外,還能夠介導WT-p53調節癌細胞干性的功能。
最近的研究[15]表明,癌細胞發生EMT后可以產生干細胞特性,這一重要發現意味著EMT和癌癥干細胞存在直接聯系,也支持了本研究的推測:p53/miR-145通路可能是PC-3細胞EMT和干性之間的一個新紐帶,今后可以從此方面入手,研究生物小分子對于腫瘤發生發展的影響。本研究結果表明,WT-p53通過部分調節miR-145以抑制PC-3的遷移、侵襲、EMT和干性;不僅如此,WT-p53的功能缺失可能會造成miR-145的表達量下降,導致前列腺癌的EMT和干性增強,從而促進骨轉移。因此,研發出針對p53/miR-145調控軸的靶向藥物,可能成為治療前列腺癌骨轉移的一種有效手段。
參考文獻
[1] Macedo F, Ladeira K, Pinho F, et al. Bone metastases: an overview[J]. Oncol Rev, 2017,11(1):321.
[2] Zhao D, Tahaney W M, Mazumdar A, et al. Molecularly targeted therapies for p53-mutant cancers[J]. Cell Mol Life Sci, 2017,74(22):4171-87.
[3] Chang C J, Chao C H, Xia W, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs[J]. Nat Cell Biol, 2011, 13(3): 317-23.
[4] Heerboth S, Housman G, Leary M, et al. EMT and tumor metastasis[J]. Clin Transl Med, 2015,4:6.
[5] Clevers H. Cancer therapy: Defining stemness[J]. Nature, 2016,534(7606):176-7.
[6] Schubert J, Brabletz T. p53 Spreads out further: suppression of EMT and stemness by activating miR-200c expression[J]. Cell Res, 2011, 21(5): 705-7.
[7] Peng X, Guo W, Liu T, et al. Identification of miRs-143 and-145 that is associated with bone metastasis of prostate cancer and involved in the regulation of EMT[J]. PLoS One, 2011, 6(5): e20341.
[8] Huang S, Guo W, Tang Y, et al. miR-143 and miR-145 inhibit stem cell characteristics of PC-3 prostate cancer cells[J]. Oncol Rep, 2012, 28(5): 1831-7.
[9] Sachdeva M, Zhu S, Wu F, et al. p53 represses c-myc through induction of the tumor suppressor miR-145[J]. Proc Natl Acad Sci U S A, 2009, 106(9): 3207-12.
[10] Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method[J]. Methods, 2001, 25(4): 402-8.
[11] Zhou Z, Flesken-Nikitin A, Nikitin A Y. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts[J]. Cancer Res, 2007, 67(12): 5683-90.
[12] Martin P, Liu Y N, Pierce R, et al. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition[J]. Am J Pathol, 2011, 179(1): 422-35.
[13] Siemens H, Jackstadt R, Hunten S, et al. miR-34 and SNAIL form a doublE-negative feedback loop to regulate epithelial-mesenchymal transitions[J]. Cell Cycle, 2011, 10(24): 4256-71.
[14] Gao P, Xing A Y, Zhou G Y, et al. The molecular mechanism of microRNA-145 to suppress invasion-metastasis cascade in gastric cancer[J]. Oncogene, 2013, 32(4): 491-501.
[15] Fantozzi A, Gruber D C, Pisarsky L, et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation[J]. Cancer Res, 2014, 74(5): 1566-75.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中老年保健(2021年3期)2021-08-22 06:50:04
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
天津醫科大學學報(2021年2期)2021-03-29 05:31:08
現代臨床醫學(2021年1期)2021-01-26 00:56:02
家庭醫學(下半月)(2020年3期)2020-05-30 12:42:02
家庭醫學(下半月)(2020年3期)2020-05-30 12:42:00
家庭醫學(下半月)(2020年3期)2020-05-30 12:42:00
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
