一種利用體外轉錄制備dsRNA的方法介紹*
2018-05-22 03:07:20劉海濤舒端陽
生物學通報 2018年6期
劉海濤 舒端陽
(1 淮南師范學院生物工程學院 安徽淮南 232038 2 廣東省梅州市豐順中學 廣東梅州 514300)
RNAi(RNA interference)是由dsRNA(double strand RNA)在細胞內被切割成siRNA(small interference RNA),從而引起的同源mRNA 特異性降解,針對性地關閉某個基因的表達,為人們研究具體某個基因的功能打開一扇窗戶,所以被廣泛用于生物學、醫學等眾多領域。然而要對某個基因進行有效的沉默需要制備與此基因mRNA 同源的dsRNA,怎樣才能獲得此dsRNA? 目前應用比較廣泛的方法有化學合成法、體外轉錄法、構建載體體內轉錄法等,本實驗通過構建一個雙啟動子的表達載體,再體外轉錄制備dsRNA。
本實驗以鱗翅目昆蟲細胞色素c 基因片段為靶基因制備dsRNA,細胞色素c(cytochrome c,cyt c)是呼吸鏈中一種重要的蛋白質,正常細胞在受到凋亡因子(例如紫外線)刺激時cyt c 就會從線粒體中釋放,激活caspase 家族蛋白酶,引起一系列的通訊級聯,最終導致細胞凋亡。若將一個正常細胞的cyt c 的表達抑制后,用紫外線處理細胞還會發生凋亡,或者凋亡程度會不會減弱?這時即可用RNAi技術沉默cyt c 基因的表達。本實驗以鱗翅目昆蟲斜紋夜蛾(Spodoptera litura)的離體細胞株Sl-1 為研究材料[1],制備其cyt c 基因的一段dsRNA。
1 實驗材料
PBS緩沖液、Sl-1細胞株、提取總RNA 的Tri-zol 試劑盒、逆轉錄酶M-MLV、DNA 連接酶、限制酶BamH I 和EcoR I、Taq 酶、RNA聚合酶、PCR 儀、帶有T7 雙啟動子的質粒Litmus28i(圖1)、微量移液器、EP 管、培養皿、4 種核糖核苷酸(NTP)、4 種脫氧核苷酸(dNTP)、冷凍離心機、焦炭酸二乙酯(DEPC)。
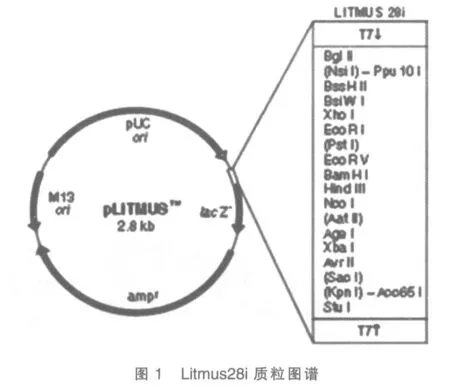
2 實驗方法
2.1 提取總RNA[2]
1)取1 瓶對數期細胞(細胞數量約1.5×106個),用PBS 洗滌后加入1 mL TRIzol;室溫靜置5 min 后移入1.5 mL 離心管中。
2)加入0.2 mL 氯仿,輕微振蕩15 s,冰上靜置2 min。
3)12000g/15 min,4℃離心后取上層水相,保留中間相和有機相(其中含有DNA 和蛋白質)。
4)加入0.5 mL 異丙醇,輕輕混勻,冰上靜置10 min。
5)12000g/10 min,4℃離心,棄上清液。
6)加入1 mL 75%乙醇,輕輕洗滌沉淀;7 500 g/5 min,4℃離心后棄上清液。
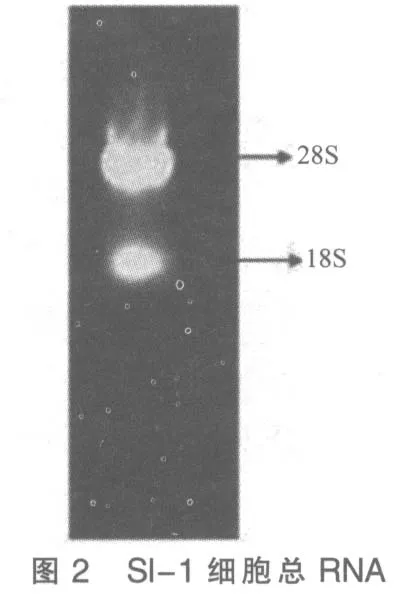
7)加入50 μL DEPC處理過的超純水溶解沉淀后取10 μL 用于瓊脂糖凝膠電泳,電泳結果顯示所提取RNA樣品中28S rRNA 亮度約為18S rRNA的2 倍,說明RNA 質量較好(如圖2所示)。
2.2 體外轉錄載體的構建
1)用微量移液器取5 μL 總RNA 為模板,加入1 μL M-MLV 逆轉錄酶、1 mg dNTP,16℃條件下對總RNA 進行逆轉錄,制備cDNA。
2)以cDNA 為模板,設計cyt c 基因片段(長度為248 bp)的引物:
上游5′-GAATTCTGCCACACTGTTGAAGCCGGT3′
下游5′-GGATCCGATCTGCACGCTCGTTTGCCT3′
引物兩頭分別加上EcoR I 和BamH I 的酶切位點,PCR 反應(圖3)。

3)PCR 產物測序正確后,電泳切膠回收。
4)37℃條件下加入EcoRⅠ和BamH Ⅰ對純化后的片段和質粒Litmus28i 同時進行酶切反應3 h。
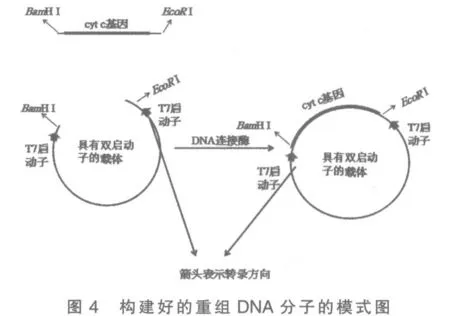
5)分別將雙酶切后的片段和質粒純化后進行連接反應,4℃,10h 得到重組DNA 分子(圖4)。
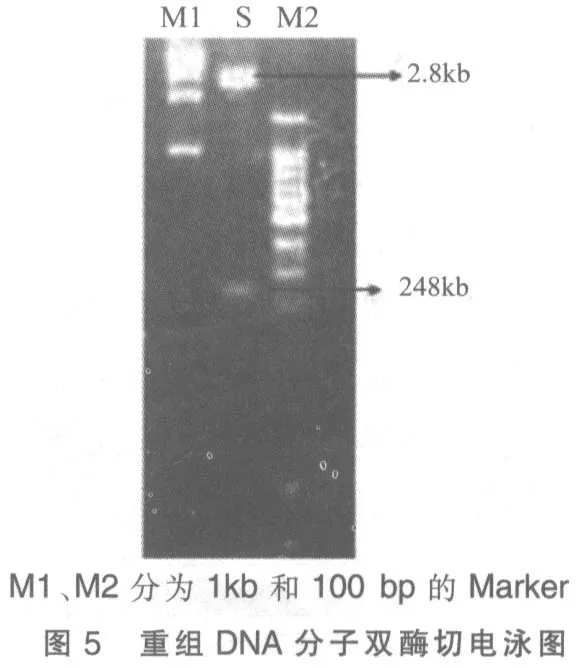
6)為保證重組DNA 分子插入正確,用EcoR Ⅰ和BamH Ⅰ同時進行雙酶切驗證,電泳檢測插入正確(圖5)。
2.3 體外轉錄
1)將重組DNA 分子分為2 份,分別用BamHⅠ和EcoR Ⅰ進行酶切,作為體外轉錄模板(圖6)。

2)加入1 μL RNA 聚合酶、NTP 2 mg 及緩沖液,37℃水浴使轉錄持續進行4 h 后,65℃加熱變性5 min 后迅速冰浴使2 種互補的單鏈RNA 退火形成dsRNA,電泳檢測(圖7)。

此時的dsRNA 序列與斜紋夜蛾cyt c 基因的一段是完全同源的,可以利用脂質體轉染Sl-1 細胞,進行RNAi 的相關實驗。
3 討論及意義
體外轉錄實驗能否成功主要取決于的限制酶選擇,只有選擇切口為5′-突出末端(5′-overhang)或平末端(blunt-end)限制酶才能成功進行體外轉錄,本實驗選用的限制酶EcoR I 和BamH I 都屬于5′-突出末端;若所采用的限制酶(如Pst I)切出的切口為3′-突出末端(3′-overhang),則RNA聚合酶在體外轉錄結束后無法與模板脫離,導致轉錄失敗。
RNA 干擾現象廣泛存在于多種生物,有效地組織外源病毒的侵染、參與機體發育、調節基因的表達等,因此研究RNAi 具有重要的意義。然而研究RNAi 的前提就是如何高效地制備相應的dsRNA。
最初研究人員是通過化學合成的方法直接合成siRNA 用于實驗,這種方法最為簡單,客戶只要知道相應目的基因的堿基序列,生物公司就會根據其要求提供高質量的siRNA,不過價格十分昂貴,且需要在靶基因中事先篩選出一段最有效的序列再進行化學合成,因此定制周期較長。
2002年,Novina CD 等[3]開發出一種利用腺病毒感染的方法將靶基因帶入受體細胞,達到了較高的轉染率且能持續穩定地在細胞內轉錄出siRNA,因此利用病毒載體是目前唯一一種可以長期研究RNAi 的方法,而且相比于化學合成這種方法的成本低。但是病毒載體只能轉染原代培養的哺乳動物細胞,對于果蠅、線蟲和斑馬魚這類生物則無能為力。
然而,通過構建表達載體在體外進行轉錄合成dsRNA 的費用相比于化學合成法要低得多,也能較快地得到dsRNA。一旦得到靶基因的模板序列,只要48 h 就可以RNAi 實驗,通常一次體外轉錄得到的dsRNA 可以進行上百次的RNAi 實驗。利用脂質體可以將dsRNA 導入受體細胞,雖然轉染效率略低于病毒載體,但是脂質體包裹著這些siRNA 可以與哺乳動物、線蟲、魚類、果蠅和各種植物的細胞發生融合,因此體外轉錄是目前進行RNAi 最有效的手段。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
兒童故事畫報(2019年5期)2019-05-26 14:26:14
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
