云南松松針藥材質量標準研究
2018-05-24 06:22:18
中國民族民間醫藥 2018年9期
楚雄醫藥高等專科學校,云南 楚雄 675000
松針(pineneedle),別名豬鬃松毛、松毛、山松須,為松科松屬植物的針狀葉。松針是我國傳統中藥,在歷代本草中均有記載。《別錄》謂其:“主風濕瘡,生毛發,安五臟”。《本草綱目》記載:“松針,氣味苦、溫、無毒,久服令人不老,輕身益氣,主風濕瘡,生毛發,安五臟,守中,不饑延年”[1]。現代研究表明,松針具有鎮痛、抗炎、鎮咳、祛痰、抗突變、降血脂、降血壓、抑菌等作用,其利用價值日益受到關注[2-4]。據文獻報道,松針中含有山柰酚等黃酮類成分[5],山柰酚在防癌、抗癌、抗感染、抗炎及免疫抑制等方面有重要作用[6]。另外,山柰酚還能夠降低血糖,改善胰島素抵抗[7]。本研究對云南松松針中所含該活性成分采用TLC及HPLC進行了定性、定量研究,并對藥材的水分、灰分、浸出物進行了測定,為制定松針的質量控制標準、開發新藥奠定了基礎。
1 儀器與材料
1.1 儀器 LC-20A高效液相色譜儀(日本島津);Sartorius BS 224S萬分之一分析天平(北京賽多利斯儀器系統有限公司);電熱鼓風干燥箱(上海-恒科學儀器有限公司);SX2-4-10箱式電阻爐(上海-恒科學儀器有限公司);索氏提取器;As10200型超聲波清洗機(北京華博科技制造有限公司);DZKW-4電子恒溫水浴鍋(上海科析試驗儀器廠)。
1.2 材料 硅膠G預制薄層板(批號20141006,青島海洋化工廠,100 mm×100 mm);HPLC級乙腈(德國MERCK公司);水為超純水;其他試劑均為分析純;山柰酚對照品(批號MUST-13121112,成都曼思特生物科技有限公司)。松針藥材9批,分別產自楚雄(2批)、南華、永仁、牟定、大姚、祿勸、大理、巍山。經楚雄醫藥高等專科學校生藥教研室姚榮林教授鑒定為云南松(PinusyunnanensisFranch)松針。
2 方法及結果
2.1 水分、總灰分、酸不溶性灰分和浸出物的測定
2.1.1 測定方法[8]水分測定參照2015年版《中華人民共和國藥典》四部0832項下水分測定法中第二法(烘干法);總灰分、酸不溶性灰分測定參照2302項下灰分測定法;浸出物測定參照2201項下浸出物測定法,以乙醇為溶劑,采用熱浸法測定醇溶性浸出物。
2.1.2 測定結果 9批樣品的水分、總灰分和酸不溶性灰分、浸出物的測定結果見表1。9批云南松松針藥材水分的平均含量為11.25%,總灰分的平均含量為3.28%,酸不溶性灰分的平均含量為0.27%,醇溶性浸出物的平均含量為20.83%。依照《中國藥典》中藥質量標準研究制定技術要求,按其平均值的±20%作為限度的制定幅度,暫定云南松松針藥材水分含量不得超過13.50%,總灰分不得超過3.94%,酸不溶性灰分不得超過0.32%,醇溶性浸出物不得少于16.66%。

表1 松針藥材各檢查項目及藥材山柰酚質量分數
2.2 松針的薄層色譜(TLC)鑒別 取云南松松針粉末2g,加甲醇-20%鹽酸(4∶1)混合溶液50 mL,回流提取1 h,濾過,濾液蒸干,殘渣加甲醇1 mL使溶解,作為供試品溶液。另取山柰酚對照品,加甲醇制成1 mL含0.5 mg的溶液,作為對照品溶液。照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗,吸取上述兩種溶液各5 μL,分別點于同一硅膠G薄層板上,以環己烷-乙酸乙酯-甲酸(4∶5∶1)為展開劑,展開,取出,晾干,噴以1%三氯化鋁顯色,在紫外燈(365 nm)下檢識,山柰酚熒光斑點清晰,分離效果理想。薄層色譜圖如圖1所示。

2.3 含量測定
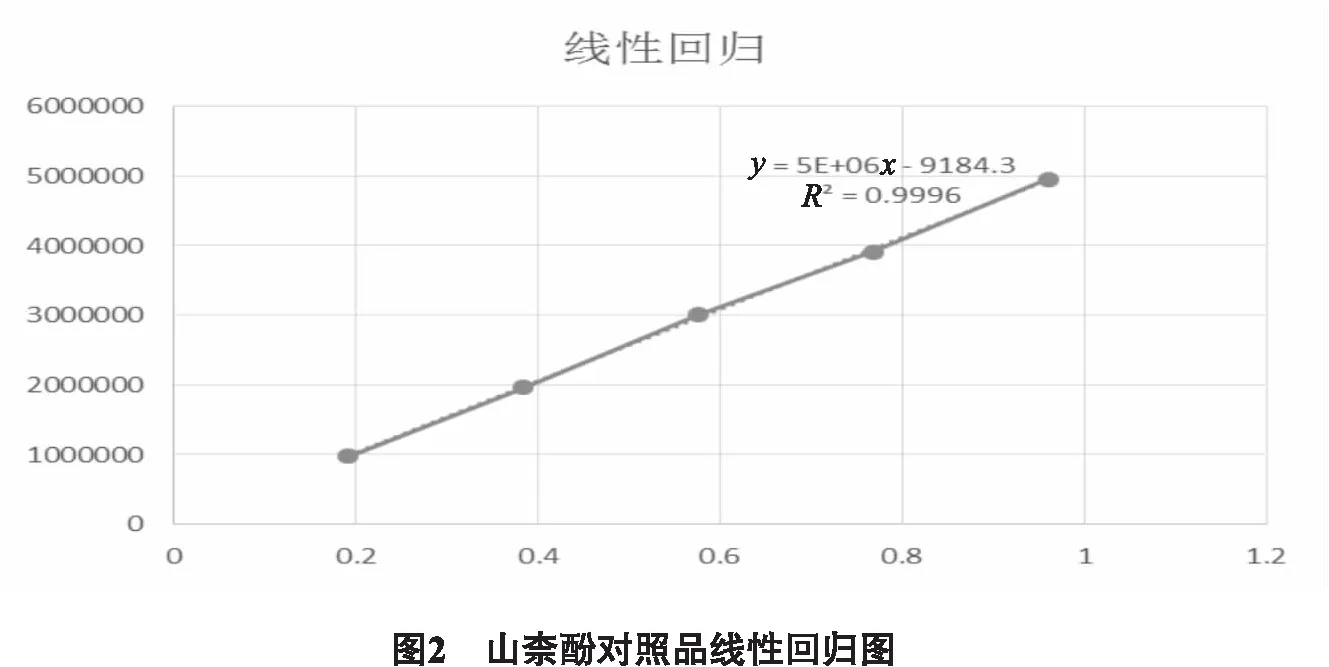
2.3.1 色譜條件 色譜柱:InertSustainC18(250 mm×4.6 mm, 5 μm);流動相:乙腈∶0.3%磷酸水溶液(30∶70);流速:1 mL/min;柱溫:40℃;檢測波長:365 nm,進樣量:10 μL。色譜柱的理論板數按山柰酚計算應不低于6000,分離度大于1.5。如圖2所示。
2.3.2 對照品溶液制備 取山柰酚對照品9 mg,精密稱定,置10 mL量瓶中,加流動相甲醇溶解并稀釋至刻度,搖勻,使成為0.9602 mg/mL的對照品儲備液。
2.3.3 供試品溶液的制備 稱取云南松松針粉末(4號篩)約1.0 g,精密稱定,各加入甲醇-20%鹽酸(4∶1)混合溶液25 mL,加熱回流提取2次,每次1.5 h,合并濾液并定容至50 mL量瓶中,用0.45 μm濾膜過濾,備用[9]。
2.3.4 線性關系考察 精密量取對照品儲備液1.0 mL置10 mL量瓶中,用流動相稀釋至刻度,搖勻,配成0.09602 mg/mL的對照品;分別精密吸取上述對照品溶液2、4、6、8、10 μL按上述色譜條件依次進樣,以山柰酚進樣量(μg)為橫坐標,峰面積(A)為縱坐標繪制標準曲線,得回歸方程為Y=5×106X-9184.3,r=0.9998,山柰酚在0.19~0.96 μg范圍內成良好的線性關系。如圖2所示。
2.3.5 精密度試驗 精密吸取山柰酚對照品溶液(0.096 02 mg/mL)10 μL,按上述色譜條件連續進樣5次,記錄山柰酚色譜峰峰面積RSD為0.84%,顯示具有良好的精密度。
2.3.6 重復性試驗 按“2.3.3”項下操作,處理同一樣品5份,精密吸取10 μL進樣檢測,記錄山柰酚的峰面積,計算RSD為1.21%(n=5),表明實驗方法的重復性較好。
2.3.7 穩定性試驗 取同一份供試液,在“2.3.1”項色譜條件下,于0、4、8、12、16 h分別記錄山柰酚峰面積,計算峰面積的RSD值為1.01%,表明供試液在16 h內穩定。
2.3.8 加樣回收率試驗 采用加樣回收法,精密稱取已知含量的藥材適量,分別精密加入一定量的山柰酚對照品,按供試品制備與測定方法,在上述色譜條件下,平行做5組,結果測得其平均回收率為101.04%,RSD為1.02%。
2.3.9 樣品測定 依上述方法,測定9批松針藥材中山柰酚含量。結果見表1。色譜圖如圖3所示,圖中1為山柰酚。

3 討論
3.1 樣品處理條件的確定 本研究考察了甲醇-10%鹽酸(4∶1)、甲醇-20%鹽酸(4∶1)和甲醇-30%鹽酸(4∶1)3種水解溶劑。結果表明以甲醇-20%鹽酸(4∶1)混合液作為水解溶劑時所得樣品含量最高,故確定采用甲醇-20%鹽酸(4∶1)混合液為水解溶劑。又以甲醇-20%鹽酸(4∶1)混合液為溶劑,比較了回流和超聲方法對提取率的影響,結果回流提取率明顯高于超聲。同時還進一步比較了不同提取時間和次數對提取效果的影響,最終確定提取2次,每次1.5 h基本能夠提取完全。
3.2 薄層條件的確定 通過比較多種展開系統苯-乙酸乙酯-乙酸、環己烷-乙酸乙酯-甲酸、甲苯-乙酸乙酯-甲酸、環己烷-三氯甲烷-丙酮等,結果表明環己烷-乙酸乙酯-甲酸(4∶5∶1)作為展開劑斑點清晰、分離度好。考察了三種不同品牌的薄層板:硅膠G薄層板(青島海洋化工廠)、硅膠G薄層板(青島美高集團有限公司)、自制硅膠G薄層板,均可對松針中山柰酚成分進行定性鑒別。按照試驗所得薄層色譜條件,考察本品在溫度15~29℃、相對濕度為40%~70%展開時,對薄層色譜結果的影響。結果表明,在上述溫度、相對濕度范圍內對該薄層色譜結果無明顯影響,9批不同產地的松針樣品均可檢出山柰酚,斑點清晰且方法簡便、快速、可行,此方法可用作松針藥材的定性鑒別。
3.3 色譜條件優化 考察了2種色譜柱,InertSustainC18(250 mm×4.6 mm, 5 μm)和Phenomenex Gemini-NX C18(250 mm×4.6 mm, 5 μm)色譜柱,結果顯示,后者所得山柰酚峰和雜質峰分離效果較差,峰形展寬,而前者分離效果較好,故選擇InertSustainC18(250 mm×4.6 mm, 5 μm)色譜柱。對不同流動相系統(甲醇-水、乙腈-1%冰醋酸、乙腈-0.3%磷酸、甲醇-0.3%磷酸水溶液),結果顯示以乙腈-0.3%磷酸水溶液(30∶70)的分離效果較好,所得峰形及峰純度最好。考察了不同流速(0.8、0.9、1.0 mL/min)、不同柱溫(25、30、40 ℃),確定最佳流速為1 mL/min,柱溫40 ℃。在上述條件下測得山柰酚相對標準偏差、分離度均符合定量測定要求。
本實驗首次比較全面建立了云南松松針藥材水分、灰分、浸出物檢查,薄層色譜鑒別以及藥材含量測定方法,且方法簡便、準確、靈敏度高,為松針藥材的質量控制提供了科學依據。
參考文獻
[1]李時珍.本草綱目[M]. 北京:人民衛生出版社,1982:1917-1923.
[2]李萍,劉友平. 松針研究進展[J]. 成都中醫藥大學學報,2001(24):49-50.
[3]胡豐林,陸瑞利.松針的利用價值分析[J].生物學雜志,1996,70(2):25-26.
[4]畢躍峰,鄭曉珂,馮衛生.松針的研究進展及開發利用[J].河南中醫藥學刊,1999,14(2):14-17.
[5]劉東彥,石曉峰,李沖,等.雪松松針醋酸乙酯部位化學成分研究[J].中草藥,2011,42(10):1921-1924.
[6]陳育華,周克元,袁漢堯.山柰酚藥效的研究進展[J].廣東醫學,2010,31(8):1064-1066.
[7]吳巧敏,金雅美,倪海祥.山柰酚對2型糖尿病大鼠慢性并發癥相關因子的影響[J].中草藥,2015,46(12):1086-1089.
[8]國家藥典委員會.中華人民共和國藥典(四部)[S].北京:中國醫藥科技出版社,2015:202.
[9]吳琳琳,王芳,茅向軍,等.青錢柳質量標準的研究[J].中成藥,2017,39(4):745-750.
