PSGL-1通過炎癥反應在小鼠鹽敏感性高血壓中的作用機制研究
2018-05-31 08:58:48毛紅亞姜曉亮楊志偉中國醫學科學院醫學實驗動物研究所北京協和醫學院比較醫學中心北京100021
中國比較醫學雜志 2018年5期
付 慧,毛紅亞,姜曉亮,劉 星,劉 雪,楊志偉(中國醫學科學院醫學實驗動物研究所,北京協和醫學院比較醫學中心,北京 100021)
高血壓是一種常見的機體病癥,威脅到世界20%~30%的人群健康,也是腦卒中、冠心病、心功能不全、腎病等人類重大疾病的主要危險因素之一。在原發性高血壓群體中鹽敏感者占50%~60%[1 - 3]。研究表明,鹽敏感性高血壓病人并發心腦血管事件的風險遠遠高于鹽耐受性高血壓,鹽敏感性高血壓已成為威脅人類健康的重要病理因素之一[4]。鹽敏感性高血壓的病理機制比較復雜,一般認為高鹽飲食導致動脈血管收縮及腎臟鈉水代謝潴留是其主要發病機制[5 - 6],但是通過舒張血管和利尿等藥物治療,仍有相當比例的高血壓患者的血壓并沒能得到有效控制[7]。即使沒有導致血壓的變化,高鹽飲食仍然增加了心血管疾病發生的風險[4]。近年來,隨著慢性炎癥反應在鹽敏感高血壓及其它心血管疾病發生發展中作用及其機制研究的進展,炎癥反應可能是高鹽飲食導致血壓升高及其靶器官損傷的重要作用機制[8]。
P-選擇素糖蛋白配體-1(PSGL-1)是表達于白細胞表面的跨膜糖化蛋白,與白細胞活化密切相關。PSGL-1與選擇素(selectin)分子的相互作用在白細胞粘附起始階段起重要作用[9 - 12],PSGL-1可以明顯提高血小板活化因子誘導的白細胞活化,與整合素等細胞因子共同作用促進白細胞粘附[13]。研究表明,PSGL-1參與調節T淋巴細胞分化過程,放大炎癥信號,是調節T淋巴細胞參與免疫反應的重要靶點[14]。白細胞釋放的TNF-α、IL-1等炎癥因子可以誘導內皮細胞和血小板內儲存的選擇素移位到細胞表面,與白細胞表面的PSGL-1結合,進一步促進白細胞粘附和炎癥反應的發生發展[15]。因此,PSGL-1在炎癥反應發生過程中發揮重要作用,但是否參與鹽敏感性高血壓的病理發展過程有待進一步研究。
為了檢測PSGL-1在鹽敏感性高血壓發生發展中的作用及其相關機制,本研究利用PSGL-1基因敲除小鼠并通過高鹽喂養,檢測小鼠血壓變化及機體中炎癥反應和腎臟靶器官損傷情況,以探索鹽敏感性高血壓及其靶器官損傷新的病理機制,為高血壓及其靶器官損傷的防治提供參考。
1 材料和方法
1.1 實驗動物
SPF級PSGL-1基因敲除(PSGL-1-/-)小鼠及其野生型(PSGL-1+/+)對照小鼠由美國俄克拉荷馬大學健康科學中心夏利軍教授捐贈,實驗動物均在中國醫學科學院醫學實驗動物研究所SPF級環境飼養[SYXK (京) 2015-0035],動物實驗方案獲實驗動物使用與管理委員會(IACUC)的批準[ILAS-PG-2014-006]。將8周齡、體重約20 g的雄性PSGL-1-/-和PSGL-1+/+小鼠各12只,分別給予高鹽(6% NaCl)和正常鹽(0.4% NaCl)喂養3個月[16],然后,在麻醉(0.6%戊巴比妥)狀態下利用頸總動脈插管法測量小鼠血壓,犧牲小鼠,取小鼠皮膚、腎臟等組織,用于炎癥細胞浸潤和相關炎癥因子表達,以及靶器官損傷的檢測。
1.2 主要試劑與儀器
IL-6單克隆抗體購自Cell Signaling Technology(#12912);TNF-α單克隆抗體購自Santa Cruz(SC-52746);IL-1β多克隆抗體購自Santa Cruz(SC-7884);CD3單克隆抗體購自Santa Cruz(SC-20047);CD68單克隆抗體購自Novus(NBP1-74570SS);CTGF多克隆抗購自Proteintech(23936-1-AP);BCA蛋白濃度測定試劑盒(增強型)購自碧云天(P0010);兔超敏二步法試劑盒購自中杉金橋(PV9001);小鼠超敏二步法試劑盒購自中杉金橋(PV9002);DAB顯色試劑盒(20 ×)購自中杉金橋(ZLI-9019);CD45-APC/Cy7小鼠抗體購自Biolegend(103116)、CD3-PE/Cy7小鼠抗體購自Biolegend(100320)。
多通道生理儀(BIOPAC Systems, Inc.);血壓檢測儀(BP-2000 Series II, Visitech Systems);電泳儀(北京六一儀器廠,DYY-C);電泳槽(上海天能,VE-180);電轉槽(上海天能,VE-186);多功能酶標儀(Multiskan GO);化學發光凝膠成像儀(Tanon-5500);正向顯微鏡(Leica DM6000 B);低溫高速離心機(Beckman);流式細胞儀(BD FACSCantoTM)。
1.3 實驗方法
1.3.1 免疫組化方法檢測組織的炎癥細胞浸潤
取小鼠皮膚組織常規固定,將石蠟包埋的切片(4 μm)二甲苯脫蠟后在不同濃度梯度酒精溶液中由高向低浸泡浸水,并使用0.3%過氧化氫阻斷內源性過氧化物酶活性。在檸檬酸緩沖液(pH 6.0)中通過微波熱誘導表位回收法揭開抗原。用羊血清阻斷非特異性結合,然后用目標單克隆抗體雜交,4℃溫育過夜。用超敏二步法試劑盒孵育后DAB顯色試劑盒鏡下顯色,蘇木素染色,鏡下觀察。
1.3.2 Western blot方法檢測腎臟組織中的蛋白表達
提取新鮮小鼠腎臟全蛋白,用BCA法檢測蛋白濃度。采用10%聚丙烯酰胺凝膠電泳,流轉至硝酸纖維素膜,封閉液(0.1%緩血酸銨緩沖液配制5%脫脂奶粉)室溫封閉2 h,加特異性抗體4℃孵育過夜,0.1%緩血酸銨緩沖液洗膜3次后加辣根過氧化物標記的IgG室溫孵育1 h,洗膜3次后,化學發光凝膠成像儀自動曝光顯影后使用Image J軟件分析灰度,結果以目的蛋白/β-actin灰度值比值表示蛋白質的相對表達水平。
1.3.3 流式細胞術檢測主動脈血管中炎癥細胞浸潤
為了進一步驗證炎癥細胞的浸潤情況,本研究剝離小鼠胸主動脈和腹主動脈血管,將其剪碎消化至單個細胞懸液后,加入流式抗體CD45-APC/Cy7、CD3-PE/Cy7,充分混勻后室溫避光孵育30 min,上機檢測小鼠主動脈血管中浸潤的白細胞和T淋巴細胞的百分率。
1.4 統計學方法
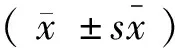
2 結果
2.1 PSGL-1參與高鹽誘導的高血壓發生發展
本研究給予PSGL-1-/-小鼠和PSGL-1+/+小鼠高鹽(6% NaCl)或正常鹽(0.4% NaCl)喂養,發現,對于PSGL-1+/+小鼠,高鹽喂養組的收縮壓、舒張壓和平均脈壓均明顯高于正常鹽喂養組(P< 0.05);而對于PSGL-1-/-小鼠,高鹽喂養組和正常鹽喂養組的血壓并無明顯變化(P> 0.05)(圖1)。提示PSGL-1在鹽敏感性高血壓的發生發展過程中起重要作用。
2.2 PSGL-1參與高鹽誘導的皮膚組織炎癥細胞浸潤的調節
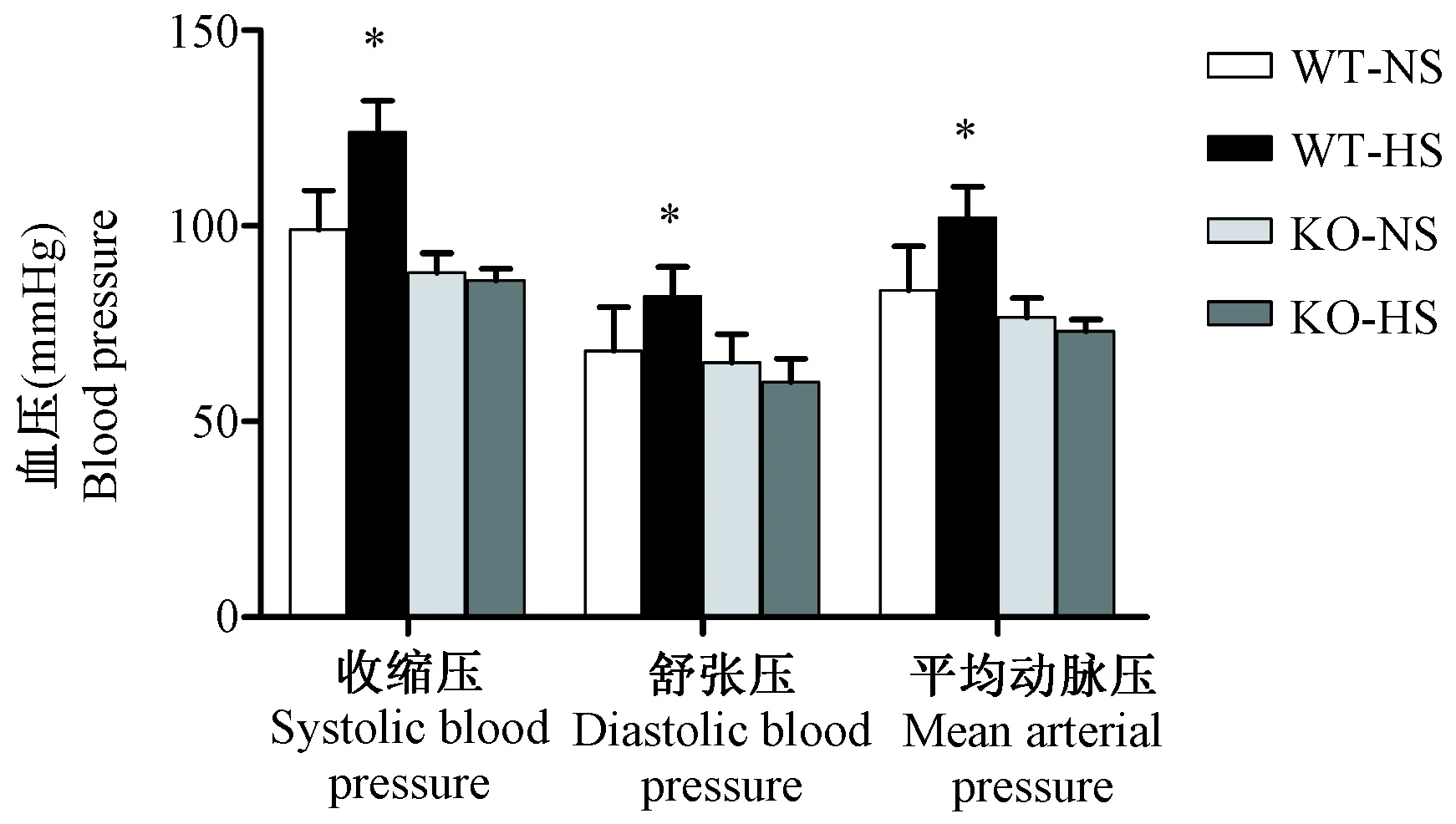
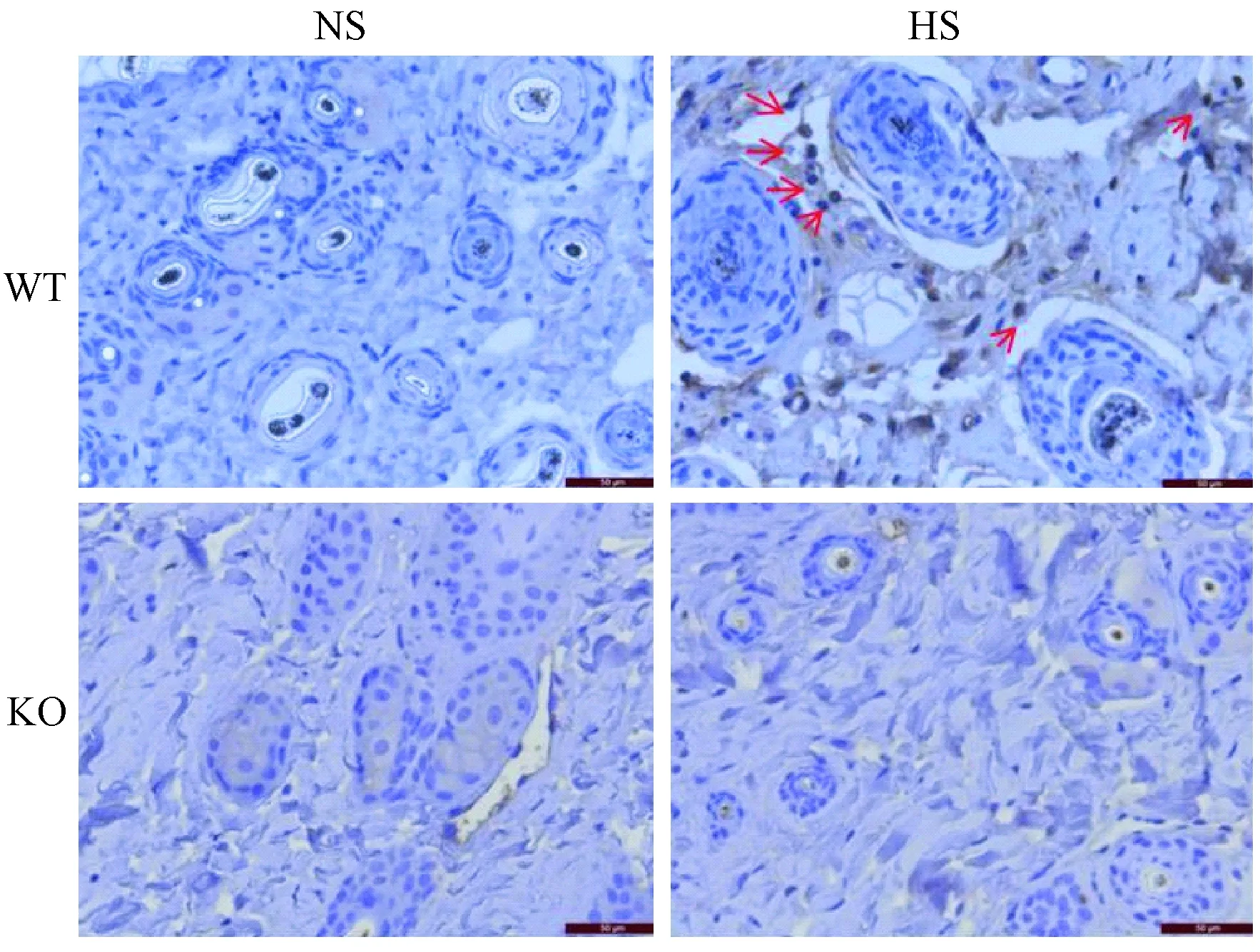
本研究用免疫組織化學染色法對小鼠皮膚組織浸潤T細胞和巨噬細胞特異性染色,發現在PSGL-1+/+小鼠高鹽喂養后皮膚組織的T細胞和巨噬細胞浸潤數量明顯增多,而PSGL-1-/-小鼠高鹽喂養后其T細胞和巨噬細胞浸潤數量并未發現明顯的變化(圖2,圖3)。表明PSGL-1在炎癥細胞浸潤過程中起重要作用,PSGL-1的缺失抑制高鹽誘導的炎癥細胞在組織中的浸潤。
注:WT:PSGL-1+/+小鼠;KO:PSGL-1-/-小鼠;NS:0.4% NaCl;HS:6% NaCl。下同。與其它組小鼠比較,one-way ANOVA,* P< 0.05。圖1 頸動脈插管法檢測PSGL-1+/+小鼠和PSGL-1-/-小鼠動脈血壓的變化Note.WT: PSGL-1+/+mice; KO: PSGL-1-/-mice; NS: 0.4% NaCl; HS: 6% NaCl. The same below. Compared with rats in the other groups, one-way ANOVA, * P< 0.05.Fig.1 Blood pressure in PSGL-1+/+ and PSGL-1-/- mice measured by carotid catheterization
注:標尺=50 μm。圖2 T淋巴細胞在PSGL-1+/+和PSGL-1-/-小鼠皮膚組織的浸潤情況Note.Bars=50 μm.Fig.2 Infiltration of T lymphocytes in the skin of PSGL-1+/+ and PSGL-1-/- mice
注:標尺=50 μm.圖3 巨噬細胞在PSGL-1+/+和PSGL-1-/-小鼠皮膚組織的浸潤情況Note.Bars=50 μm.Fig.3 Infiltration of macrophages in the skin of PSGL-1+/+ and PSGL-1-/- mice
2.3 PSGL-1參與高鹽誘導的主動脈血管中炎癥細胞浸潤的調節
用流式細胞術檢測小鼠主動脈血管中炎癥細胞浸潤數量,發現在PSGL-1+/+小鼠高鹽喂養后小鼠主動脈血管中CD45+白細胞和CD3e+T細胞的浸潤數量明顯增多,而PSGL-1-/-小鼠高鹽喂養后其浸潤數量并沒有出現明顯變化(圖4A,4B),證明PSGL-1參與高鹽誘導的炎癥細胞向主動脈中浸潤的過程。PSGL-1缺失對此過程有抑制作用,從而抑制炎癥反應的發生發展。
2.4 PSGL-1參與高鹽誘導的腎臟炎癥因子表達的調節
腎臟是機體血壓調節的重要器官,為了進一步驗證機體炎癥反應發生情況,本研究檢測了腎臟IL-6、IL-1β、TNF-α蛋白水平的表達。檢測結果發現,PSGL-1+/+小鼠,高鹽喂養組腎臟IL-6、IL-1β、TNF-α的表達明顯高于正常鹽喂養組;而PSGL-1-/-小鼠,高鹽喂養組與正常鹽喂養組腎臟IL-6、IL-1β、TNF-α表達無明顯差異(圖5A,5B,5C)。表明PSGL-1基因缺失對高鹽誘導的腎臟炎癥反應有抑制作用。
2.5 PSGL-1參與高鹽誘導的腎臟纖維化水平的調節
為了檢測高鹽對腎臟損傷的影響,本研究檢測了腎臟中作為腎臟損傷的標示蛋白結締組織生長因子(connective tissue growth factor,CTGF)[17]的表達情況。檢測發現,PSGL-1+/+小鼠高鹽喂養組腎臟CTGF蛋白表達量明顯高于正常鹽喂養組;而PSGL-1-/-小鼠腎臟中,高鹽喂養組CTGF蛋白的表達與正常鹽喂養組無明顯差異(圖6)。結果表明,PSGL-1基因缺失對高鹽誘導的腎臟纖維化具有抑制作用。
3 討論
高鹽攝入是鹽敏感性高血壓發生發展的重要因素,鹽敏感性高血壓發生發展的機理主要包括腎臟對鈉水代謝調節、血管平滑肌的收縮、交感神經系統興奮等[5, 18 - 19]。近年來,鹽敏感性高血壓被認為是一種低度炎癥性疾病,免疫系統在鹽敏感性高血壓的發生發展中的作用引起研究者的重視。高鹽等環境刺激下,作為與選擇素(selectin)結合的配體,白細胞表面的P-選擇素糖蛋白配體-1(PSGL-1)識別并結合表達在活化的內皮細胞表面的選擇素,促進白細胞在血管內皮的粘附和浸潤[9 - 10, 12, 20],促進炎癥反應的發生發展。
本研究發現,通過高鹽飲食誘發野生型小鼠發生鹽敏感性高血壓,且其麻醉狀態下血壓升高值與其它小鼠鹽敏感高血壓模型的血壓升高值相近,如去氧皮質酮(deoxycorticosterone,DOCA)緩釋誘發小鼠鹽敏感性高血壓模型[21],血管緊張素II誘導的高血壓模型[22]等。人類收縮壓低于120 mmHg時為理想血壓,高于140 mmHg時為高血壓[23],其差值與本實驗中血壓升高值相近。另外野生型小鼠高鹽飲食后動脈血管和皮膚中炎癥細胞(T細胞、巨噬細胞)浸潤、炎癥因子(IL-6、IL-1β、TNF-α)及腎臟纖維化標識蛋白(結締組織生長因子,CTGF)表達增加,而PSGL-1敲除后小鼠并未發生以上現象。該結果提示PSGL-1可能通過炎癥反應參與了鹽敏感性高血壓的發生發展及其靶器官的損傷。
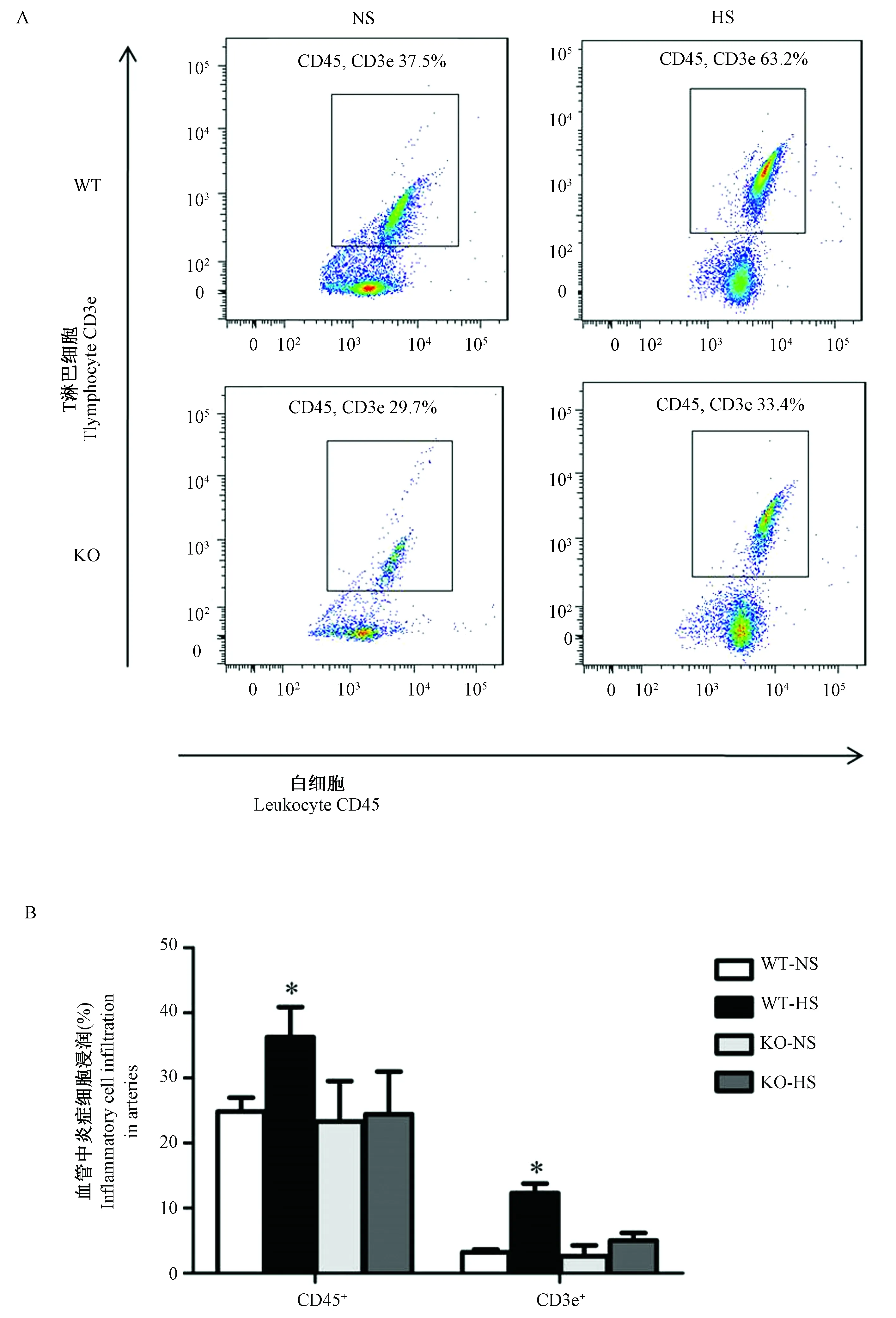
注:A:主動脈血管中浸潤的炎癥細胞圖譜(流式細胞術散點圖)。B:浸潤的炎癥細胞在主動脈血管細胞中的比例。與其它組小鼠比較,one-way ANOVA,* P< 0.05。圖4 白細胞和T淋巴細胞在PSGL-1+/+和PSGL-1-/-小鼠主動脈血管中的浸潤Note.A: Inflammatory cells in the aorta (flow cytometry scatter plot). B: Percentage of infiltrating inflammatory cells in aortic vascular cells. Compared with rats in the other groups, one-way ANOVA,* P< 0.05.Fig.4 Infiltration of leukocytes and T lymphocytes in the aorta of PSGL-1+/+ and PSGL-1-/- mice
有研究表明,高鹽誘導Dahl鹽敏感大鼠發生高血壓同時,腎臟浸潤性T淋巴細胞和巨噬細胞顯著增加[11],腎臟中浸潤的T淋巴細胞和巨噬細胞等炎癥細胞分泌多種細胞因子[11, 24],直接或間接參與了鹽敏感性高血壓的發生發展和靶器官損傷[25 - 26]。IL-6、IL-1β和TNF-α均是炎癥反應發生發展過程中發揮重要作用的炎癥因子或促炎因子[27]。IL-6可通過進一步介導巨噬細胞在腎臟中的浸潤增加或增殖[11, 28 - 29];IL-1β經炎癥小體導致腎臟炎癥和腎臟纖維化[30];IL-17激活RhoA/Rho激酶促進內皮功能障礙等參與高血壓及靶器官損傷;TNF-α作用于血管內皮細胞促使其分泌其他炎癥因子促進炎癥反應[29],同時通過抑制eNOS表達以抑制血管舒張作用,或通過調節腎臟的血液動力學和排泄功能[31],促進高血壓及靶器官損傷的發生發展[11, 32]。本研究發現,高鹽喂養小鼠的血壓明顯升高,并伴隨其皮膚T細胞、巨噬細胞浸潤增多和腎臟組織中IL-6、IL-1β、TNF-α表達量升高,而PSGL-1缺陷小鼠高鹽喂養后并未引起其血壓、炎癥細胞浸潤和腎臟組織炎癥因子表達量的明顯變化,顯示PSGL-1可能通過炎癥反應參與鹽敏感性高血壓的發生發展,但具體的作用機制還有待于進一步研究。
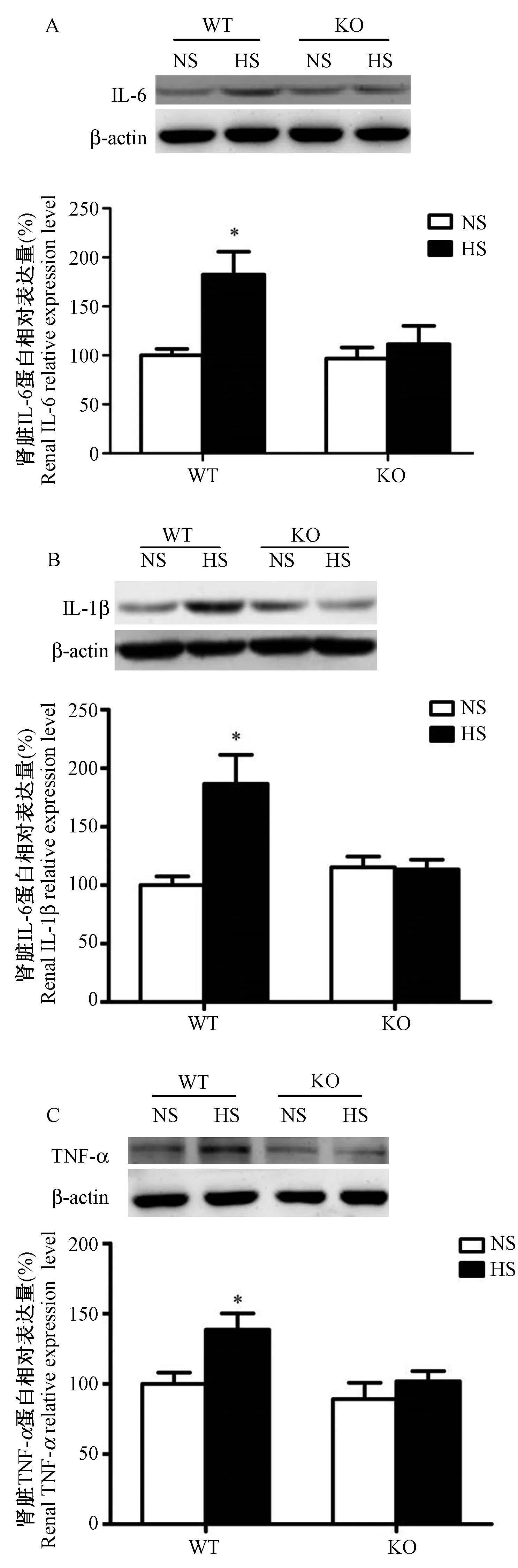
注:A、B、C:分別為小鼠腎臟中IL-6、IL-1β、TGF-α蛋白的相對表達量。與其它組小鼠比較,one-way ANOVA,* P< 0.05。圖5 PSGL-1+/+和PSGL-1-/-小鼠腎臟中IL-6、IL-1β和TNF-α蛋白表達Note.A, B, C: The expression of IL-6, IL-1β, TGF-α in the kidney of PSGL-1+/+ and PSGL-1-/- mice, respectively. Compared with rats in the other groups, one-way ANOVA, * P< 0.05.Fig.5 Interleukin-6, interleukin-1β, and tumor necrosis factor-α expression in the kidney of PSGL-1+/+ and PSGL-1-/- mice
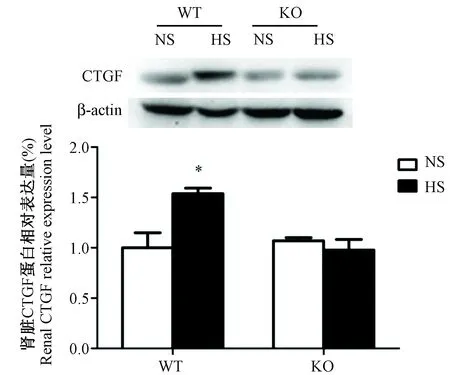
注:與其它組小鼠比較,one-way ANOVA,* P< 0.05。圖6 PSGL-1+/+和PSGL-1-/-小鼠腎臟中CTGF蛋白的表達Note.Compared with rats in the other groups, one-way ANOVA,* P< 0.05.Fig.6 Connective tissue growth factor expression in the kidney of PSGL-1+/+ and PSGL-1-/- mice
結締組織生長因子CTGF是一種炎癥介質,CTGF的表達受炎癥因子(如TNF-α,IL-1β,TGF-β等)及其它細胞外基質酶等多種炎癥介質的調節[33],腎臟在發生損傷時CTGF表達升高。作為促纖維化因子TGF-β的下游信號介質,CTGF表達增加可能是器官纖維化發生發展的中心環節,CTGF與TGF-β結合增強TGF-β與其受體的二聚化[34],進一步促進TGF-β信號傳導,促進組織纖維化,導致腎臟功能損傷[35]。CTGF能通過激活αvβ5整合素,凋亡信號調節激酶1,p38/JNK和AP-1/NF-κB通路,上調IL-6的表達,促進炎癥反應[36]。本研究發現,在高鹽喂養野生型小鼠發生高血壓的同時,腎臟中CTGF的表達量明顯增多,而PSGL-1缺失小鼠高鹽喂養后血壓和CTGF的表達量都無明顯變化,表明PSGL-1缺失能夠抑制高鹽誘導的組織炎癥反應發生而導致的血壓升高和腎臟器官損傷。但是,高鹽誘導的高血壓和腎臟損傷之間的因果關系還有待進一步研究。
高血壓控制的根本目的是靶器官損傷的預防與治療。本研究揭示了PSGL-1通過調節炎癥反應在高鹽誘導的鹽敏感性高血壓的發生發展和靶器官損傷中的重要作用,并初步探索了PSGL-1通過調控T細胞和巨噬細胞浸潤參與鹽敏感性高血壓的可能機制,為高血壓的預防和治療提供了新的理論依據。
參考文獻:
[1] Franco V, Oparil S. Salt sensitivity, a determinant of blood pressure, cardiovascular disease and survival [J]. J Am Coll Nutr, 2006, 25(3 Suppl): 247S - 255S.
[2] Meneton P, Jeunemaitre X, de Wardener HE, et al. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases [J]. Physiol Rev, 2005, 85(2): 679 - 715.
[3] Weinberger MH, Fineberg NS, Fineberg SE, et al. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans [J]. Hypertension, 2001, 37(2 Pt 2): 429 - 432.
[4] Laffer CL, Scott RR, Titze JM, et al. Hemodynamics and salt-and-water balance link sodium storage and vascular dysfunction in salt-sensitive subjects [J]. Hypertension, 2016, 68(1): 195 - 203.
[5] Iwamoto T, Kita S, Zhang J, et al. Salt-sensitive hypertension is triggered by Ca2+entry via Na+/Ca2+exchanger type-1 in vascular smooth muscle [J]. Nat Med, 2004, 10(11): 1193 - 1199.
[6] Machnik A, Neuhofer W, Jantsch J, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism [J]. Nat Med, 2009, 15(5): 545 - 552.
[7] Guessous I, Bochud M, Theler JM, et al. 1999-2009 Trends in prevalence, unawareness, treatment and control of hypertension in Geneva, Switzerland [J]. PLoS One, 2012, 7(6): e39877.
[8] Schiffrin EL. Mechanisms of remodelling of small arteries, antihypertensive therapy and the immune system in hypertension [J]. Clin Invest Med, 2015, 38(6): E394 - E402.
[9] Moore KL, Stults NL, Diaz S, et al. Identification of a specific glycoprotein ligand for P-selectin (CD62) on myeloid cells [J]. J Cell Biol, 1992, 118(2): 445 - 456.
[10] Sako D, Chang XJ, Barone KM, et al. Expression cloning of a functional glycoprotein ligand for P-selectin [J]. Cell, 1993, 75(6): 1179 - 1186.
[11] Rudemiller N, Lund H, Jacob HJ, et al. CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney [J]. Hypertension, 2014, 63(3): 559 - 564.
[12] Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response [J]. Lab Invest, 2002, 82(5): 521 - 533.
[13] 曾憲錄, 王曉光, 高艷光, 等. PSGL-1——一種介導白細胞粘附的重要分子 [J]. 分子科學學報, 2002, 18(2): 84 - 89.
[14] Tinoco R, Otero DC, Takahashi AA, et al. PSGL-1: a new player in the immune checkpoint landscape [J]. Trends Immunol, 2017, 38(5): 323 - 335.
[15] Frenette PS, Denis CV, Weiss L, et al. P-Selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactionsinvivo[J]. J Exp Med, 2000, 191(8): 1413 - 1422.
[16] Chen L, Song H, Wang Y, et al. Arterial α2-Na+pump expression influences blood pressure: lessons from novel, genetically engineered smooth muscle-specific α2 mice [J]. Am J Physiol Heart Circ Physiol, 2015, 309(5): H958 - H968.
[17] Mason RM. Fell-Muir lecture: Connective tissue growth factor (CCN2) — a pernicious and pleiotropic player in the development of kidney fibrosis [J]. Int J Exp Pathol, 2013, 94(1): 1 - 16.
[18] Jose PA, Yang Z, Zeng C, et al. The importance of the gastrorenal axis in the control of body sodium homeostasis [J]. Exp Physiol, 2016, 101(4): 465 - 470.
[19] Fujita T. Mechanism of salt-sensitive hypertension: focus on adrenal and sympathetic nervous systems [J]. J Am Soc Nephrol, 2014, 25(6): 1148 - 1155.
[20] Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response [J]. Lab Invest, 2002, 82(5): 521 - 533.
[21] Grobe JL, Buehrer BA, Hilzendeger AM, et al. Angiotensinergic signaling in the brain mediates metabolic effects of deoxycorticosterone (DOCA)-salt in C57 mice [J]. Hypertension, 2011, 57(3): 600 - 607.
[22] Kirabo A, Fontana V, de Faria AP, et al. DC isoketal-modified proteins activate T cells and promote hypertension [J]. J Clin Invest, 2014, 124(10): 4642 - 4656.
[23] 劉力生. 中國高血壓防治指南2010 [J]. 中國醫學前沿雜志(電子版), 2011, 3(5): 42 - 93.
[24] Thang LV, Demel SL, Crawford R, et al. Macrophage depletion lowers blood pressure and restores sympathetic nerve α2-adrenergic receptor function in mesenteric arteries of DOCA-salt hypertensive rats [J]. Am J Physiol Heart Circ Physiol, 2015, 309(7): H1186 - H1197.
[25] Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease [J]. Biochem Pharmacol, 2009, 78(6): 539 - 552.
[26] Dmitrieva NI, Burg MB. Secretion of von Willebrand factor by endothelial cells links sodium to hypercoagulability and thrombosis [J]. Proc Natl Acad Sci U S A, 2014, 111(17): 6485 - 6490.
[27] Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response [J]. Lab Invest, 2002, 82(5): 521 - 533.
[28] 趙譞, 楊新春, 蔡軍. T細胞免疫與高血壓的研究進展 [J]. 心血管病學進展, 2012, 33(1): 28 - 31.
[29] Krishnan SM, Dowling JK, Ling YH, et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice [J]. Br J Pharmacol, 2016, 173(4): 752 - 765.
[30] Mehaffey E, Majid D. Tumor necrosis factor-α, kidney function, and hypertension [J]. Am J Physiol Renal Physiol, 2017, 313(4): F1005 - F1008.
[31] Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury [J]. Am J Physiol Renal Physiol, 2014, 307(5): F499 - F508.
[32] Ramseyer VD, Garvin JL. Tumor necrosis factor-α: regulation of renal function and blood pressure [J]. Am J Physiol Renal Physiol, 2013, 304(10): F1231 - F1242.
[33] Kular L, Pakradouni J, Kitabgi P, et al. The CCN family: a new class of inflammation modulators? [J]. Biochimie, 2011, 93(3): 377 - 388.
[34] Abreu JG, Ketpura NI, Reversade B, et al. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β [J]. Nat Cell Biol, 2002, 4(8): 599 - 604.
[35] 黃平, 錢康. 絞股藍總皂苷對糖尿病腎病大鼠結締組織生長因子表達的影響 [J]. 中華中醫藥學刊, 2007, 25(11): 2235 - 2238.
[36] Liu SC, Hsu CJ, Chen HT, et al. Correction: CTGF increases IL-6 expression in human synovial fibroblasts through integrin-dependent signaling pathway [J]. PLoS One, 2015, 10(12): e144569.
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
西部醫學(2021年10期)2021-10-28 08:25:50
中老年保健(2021年5期)2021-08-24 07:07:16
學苑創造·A版(2020年9期)2020-10-13 09:41:02
中國生殖健康(2020年6期)2020-02-01 06:29:06
基層中醫藥(2018年4期)2018-08-29 01:25:58
基層中醫藥(2018年6期)2018-08-29 01:20:14
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
中國中醫藥現代遠程教育(2014年23期)2014-03-01 04:33:55
