各Li吸附組分下硅烯氫存儲性能的第一性原理研究?
2018-06-14 06:31:16盛喆戴顯英苗東銘吳淑靜趙天龍郝躍
物理學報 2018年10期
關鍵詞:結構
盛喆 戴顯英 苗東銘 吳淑靜 趙天龍 郝躍
(西安電子科技大學微電子學院,寬帶隙半導體技術國家重點學科實驗室,西安 710071)
1 引 言
作為一種重要的類石墨烯二維材料,硅烯憑借與硅基半導體技術兼容的獨特優勢,在電子器件[1?3]、傳感器[4]、催化劑[5]等領域受到密切關注.近年來,隨著各國對氫能源的不斷使用和重視,科研工作者開始利用硅烯較低的分子量和高比表面積的獨特優勢[6,7],研究硅烯在常溫常壓環境下作為氫存儲材料的潛在可能性[8?10],從而避免使用傳統高成本、高風險和較低存儲密度的液化和加壓的氫存儲方式[9,11,12].研究發現,純硅烯儲氫能力很弱[13],吸附一個氫氣分子的平均吸附能只有0.11 eV/H2,遠小于美國能源部(U.S.Department of Energy,DOE)提出的5.50 wt%質量儲氫密度和0.20—0.60 eV/H2平均吸附能的氫存儲標準[6,9,13,14].但是由于硅烯具有sp2/sp3混合雜化的特性[15,16],在合適的金屬修飾后,硅烯能夠通過π鍵和金屬原子緊密結合,并以金屬原子為吸附中心吸附較多的氫氣分子.這些金屬原子也不會因為聚合而形成團簇[9,17],能夠穩定分布在硅烯表面為氫氣分子提供持久而有效的吸附環境,這一點對于金屬修飾硅烯在氫存儲領域中的循環使用尤為重要[11,18?20].
進一步研究發現,以Li,Na為代表的堿金屬由于具備較低的內聚能[9,10,17]、較小的分子量[6,21]以及能夠提供較為適宜的氫氣吸附強度(介于強化學吸附和弱物理吸附之間)[22],比較適合用于氫存儲領域中硅烯的修飾.Wang等[9]基于第一性原理計算,發現Li修飾硅烯(Li組分為0.20)中Li的結合能(2.69 eV/Li)遠遠大于其內聚能(1.71 eV),因此Li原子能夠在硅烯表面穩定分布;進一步計算得到Li修飾硅烯的最大質量儲氫密度為4.82 wt%,相應的平均吸附能為0.21 eV/H2,略低于DOE的氫存儲標準,但也強于純硅烯.Hussain等[22]利用第一性原理方法對Na修飾硅烯(Na組分為0.20)的儲氫能力進行了研究,發現Na原子在硅烯表面也具有較大的結合能,并且每個Na原子能夠吸附5個氫氣分子,最終能夠得到6.90 wt%的質量儲氫密度和0.48 eV/H2的平均吸附能.顯然,堿金屬修飾硅烯能夠有效提高硅烯的氫存儲能力.但是,絕大部分科研工作者只是對單一組分下堿金屬修飾硅烯的氫存儲性能進行了研究,未曾探究不同組分堿金屬對硅烯氫存儲性能的影響,而后者可能對充分挖掘堿金屬修飾硅烯的氫存儲能力具有重要意義,并可為今后硅烯在氫存儲領域的應用提供理論參考.
因此,基于第一性原理計算方法,本文從Li修飾硅烯入手,研究了不同Li吸附組分下硅烯體系(LixSi1?x)的結構和穩定性,討論了各Li吸附組分下硅烯的儲氫能力,并以質量儲氫密度最高的Li飽和吸附硅烯為例,進一步討論了Li修飾硅烯的儲氫機理.結果表明,提高Li組分甚至使其達到飽和在理論上能使Li修飾硅烯具備更強的儲氫能力,且電荷轉移誘導的靜電相互作用和軌道雜化作用是Li修飾硅烯儲氫的關鍵.
2 計算方法
采用DMol3程序包[23]對硅烯進行第一性原理計算.其中,交換關聯函數選擇廣義梯度近似(generalized gradient approximation,GGA)[24]下的Perdew-Burke-Ernzerhof泛函[25],基函數選擇的是包含軌道極化函數的雙數值軌道基組.此外,選用Grimme[26]提出的半經驗DFT-D2方法修正范德瓦耳斯(van der Waals,vdW)相互作用,以獲得較為可靠的庫侖作用和氫氣吸附能.計算過程中采用8×8×1的K點網格[27],5.1 ?的軌道截斷半徑.結構優化過程中,原子相互作用力閾值設為0.001 Ha/?,原子移動的最大距離閾值設為3.0×10?3?,體系中每個原子的能量收斂精度設為1.0×10?6Ha/atom.此外,為了研究各Li修飾硅烯體系中Li原子和硅烯表面的結合能力以及各Li修飾硅烯對氫氣分子的吸附能力,定義Li原子的平均結合能(Eb)和氫氣分子的平均吸附能(Eads)為:

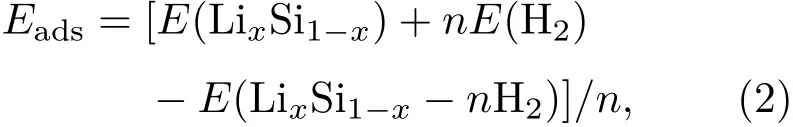
其中E(silicene)和E(LixSi1?x)分別表示純硅烯在m個Li原子吸附修飾前、后的總能量,x代表這m個Li原子所對應的組分大小;E(LixSi1?x?nH2)表示吸附n個氫氣分子后Li原子修飾硅烯的總能量;E(Li)和E(H2)則分別表示相同晶胞尺寸下單個孤立Li原子以及單個孤立氫氣分子的能量;m和n分別表示Li原子和氫氣分子的數量.
3 結果與討論
3.1 純硅烯結構模型
硅烯是類石墨烯的二維薄膜材料,與石墨烯不同的是,其結構中具有2套不在同一平面的子晶格,因此組成翹曲的蜂窩狀晶格結構,如圖1所示.經結構優化,得到的原胞晶格常數為3.87 ?,最近鄰Si原子之間鍵長為2.28 ?,兩套晶格之間的垂直距離差(翹曲高度)h=0.46 ?,各參數結果和前人的報道相符合[6,7,13,22].為了最大化Li修飾硅烯的儲氫量,選用單層2×2硅烯超胞(含8個Si原子)作為氫存儲的研究對象,并且在考慮精度和效率的前提下,使用20 ?的真空層,以消除z軸方向上由周期性帶來的影響.
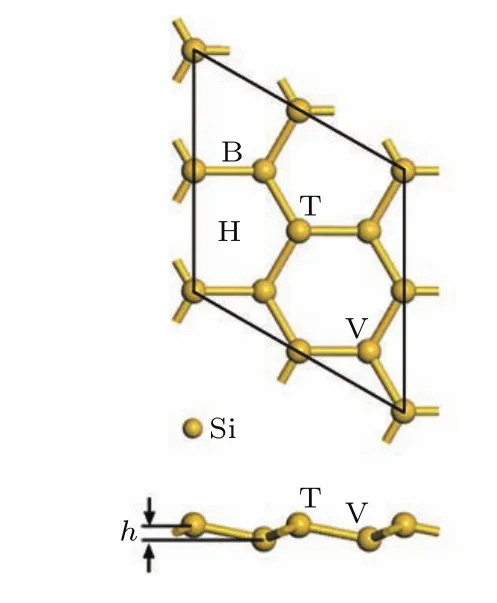
圖1 2×2單層硅烯超胞優化結構俯視和側視圖Fig.1.Top(upper panel)and side(bottom panel)views of the geometry structure of 2×2 supercell silicene.
3.2 不同Li組分下硅烯體系的結構優化及分析
3.2.1 優化結構分析
對于單個Li原子而言,在硅烯表面有四種典型的吸附位置,如圖1中所示,分別為頂位(T)、谷位(V)、橋位(B)以及六元環中空位(H).先將1個Li原子分別放置在硅烯超胞表面的四種典型位置上進行結構優化,優化后的硅烯結構和對應的Li原子平均結合能分別如圖2和圖3所示.從圖中可知,單個Li原子吸附在中空位時該硅烯體系(Li0.11Si0.89硅烯)具有最低的能量,相應的Li原子的平均結合能為2.11 eV/Li,說明中空位是單個Li原子在硅烯表面吸附的最穩定位置.值得注意的是,單個Li原子的平均結合能大于Li原子之間的內聚能(1.71 eV),可見該Li原子和硅烯之間存在較強的結合,能使Li0.11Si0.89硅烯即使在多次吸附和釋放氫氣后依然保持穩定.

圖2 不同Li組分下硅烯體系的結構俯視和側視圖Fig.2.Top(upper panels)and side(bottom panels)views of geometry structures of LixSi1?x.
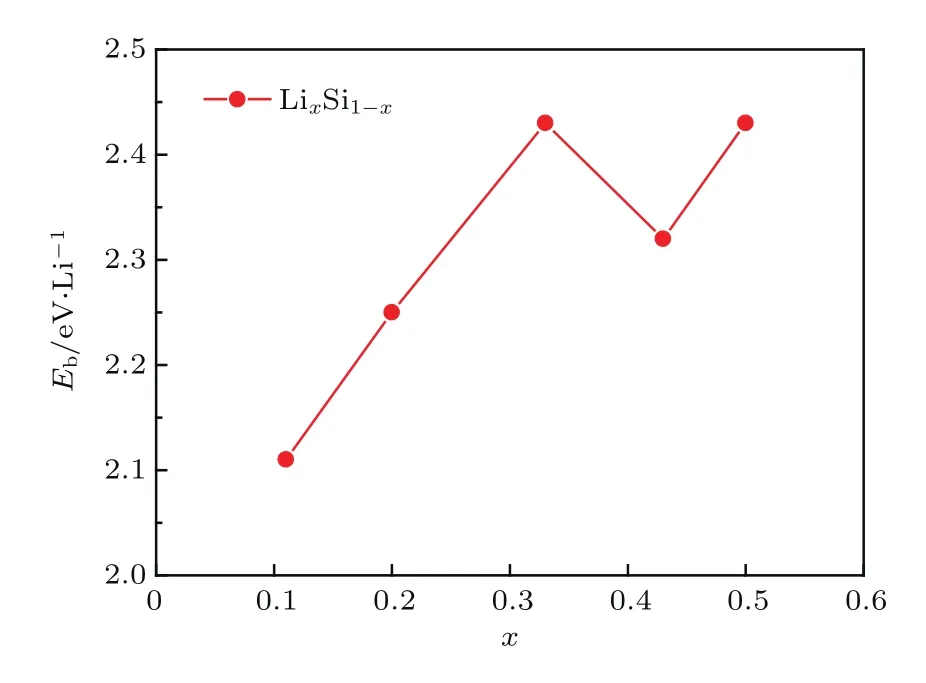
圖3 不同Li組分硅烯體系中Li原子的平均結合能Fig.3.Binding energies of Li in LixSi1?x.
依次將2個、4個、6個和8個Li原子(分別對應Li0.20Si0.80,Li0.33Si0.67,Li0.43Si0.57和Li0.50Si0.50硅烯)放置在硅烯超胞表面各吸附位置并進行結構優化.從圖2和圖3中可以看到,Li0.20Si0.80硅烯中Li原子的最穩定位置依然是中空位,且超胞中的兩個Li原子分別位于硅烯的上下表面,對應的平均結合能為2.25 eV/Li.在更高的Li組分下(Li0.33Si0.67硅烯),Li原子的最穩定位置轉變到了谷位和橋位之間,并且具有2.43 eV/Li的平均結合能.隨著Li組分的繼續增加(Li0.43Si0.57和Li0.50Si0.50硅烯),Li原子的穩定位置逐步轉向谷位,所對應的平均結合能分別為2.33 eV/Li和2.43 eV/Li.繼續增大Li吸附組分,結構優化后發現Li原子的平均結合能大幅減小,并有低于其內聚能的趨勢,因此本文認為當Li吸附組分為0.50時,Li原子在硅烯表面的吸附達到飽和,這也與前人的報道相符合[28,29].總體來看,隨著Li組分從0.11增加到0.50,Li原子的平均結合能具有增大的趨勢,且都能大于其內聚能,說明Li吸附組分在其達到飽和前的提高并不會給Li修飾硅烯的穩定性造成較大影響,后續對各結構進行氫存儲研究是有意義的.
3.2.2 結合機理分析
為了理解Li原子和硅烯的結合機理,本文以Li吸附組分最高的Li0.50Si0.50硅烯結構為例,進行了Mulliken電荷布居和態密度分析.Mulliken電荷布居顯示,由于Si原子相較于Li原子具有更大的電負性,每個Li原子向硅烯轉移了約0.77個電子,從而使Li和Si原子分別變為帶正電和負電的離子.帶正電的Li離子和帶負電的Si離子會形成一個較強的局域電場,這對于后續的氫氣存儲非常重要.同時,從圖4的態密度圖中可以看到,Si的s和p軌道與Li的s和p軌道產生了顯著的雜化(?4.98—?0.51 eV以及0.60—6.23 eV),表明Li的s和p軌道參與了成鍵過程,同時也暗示了Li和Si之間存在著電荷轉移.Li原子的p軌道也參與了成鍵,這是因為在Li原子和Si原子軌道雜化的過程中,Li原子首先向Si原子貢獻了自己的s電子,導致部分Si原子的p軌道被占據;隨后,周圍Si原子產生的配位場使空的Li原子p軌道發生劈裂,并將一部分電子貢獻回Li的p軌道,最終使得Li和Si之間存在較強的s-p和p-p軌道雜化[30].此外,由于Li原子的存在,Li0.50Si0.50硅烯結構產生了約0.7 eV的能隙,因此該結構具有半導體特性.

圖4 Li0.50Si0.50硅烯結構態密度圖Fig.4.Diagram of density of states of Li0.50Si0.50.
3.3 不同Li組分下硅烯體系的氫存儲研究
3.3.1 最大儲氫量判斷和結構分析
基于上述優化后的硅烯結構,對各Li組分下硅烯超胞結構的氫存儲性能進行了研究.我們將氫氣分子以不同的位置和方向逐個添加在Li原子周圍,借助結構優化來判斷該硅烯體系的最大儲氫量.通常情況下,Li修飾硅烯一旦達到了最大儲氫量,后續添加的氫氣分子在結構優化后會被排斥而遠離Li原子,以保證硅烯體系的總能量最低[20].因此,定義氫氣吸附距離為氫鍵中心點到Li原子之間的距離,并通過比較各氫氣分子和第一個吸附在該Li原子上的氫氣分子的吸附距離來判斷硅烯體系是否已經達到最大儲氫量.本文以Li組分最低的Li0.11Si0.89硅烯結構為例,介紹硅烯的最大儲氫量判定過程(圖5(a)).在這一過程中,我們還統計了該硅烯結構在吸附氫氣過程中各氫氣分子的吸附距離,如圖5(b)所示.從圖5(a)和圖5(b)中可以看到,只添加了1個氫氣分子時,該氫氣分子在結構優化后傾斜地吸附在Li原子上,相應的吸附距離為1.96 ?;當添加了2個氫氣分子時,結構優化后發現,這2個氫氣分子在吸附后組成了一個俯視圖上近似線性的結構,相應的吸附距離分別為2.01 ?和2.02 ?;當添加3個氫氣分子時,各氫氣分子的吸附距離在結構優化后增加到了2.05 ?,最終組成一個俯視圖上近似對稱的三角結構;繼續添加氫氣分子,結構優化后發現添加的氫氣分子被排斥而遠離Li原子,相應的吸附距離變為3.96 ?,遠大于第一個氫氣分子的吸附距離.可見,硅烯中的單個Li原子最多能吸附3個氫氣分子,這與Wang等[9]的研究結果相同.因此,Li0.11Si0.89硅烯結構的最大質量儲氫密度為2.54 wt%,相應的平均吸附能為0.58 eV/H2.

圖5 Li0.11Si0.89硅烯最大儲氫量判斷的(a)結構圖,(b)氫氣吸附距離統計圖Fig.5.Diagrams of(a)geometry structures with(b)corresponding adsorption distance statistics of Li0.11Si0.89 during the maximum hydrogen storage judgment.
根據上述最大儲氫量判定方法,本文繼續對其他硅烯結構的最大儲氫量進行了研究.由于硅烯中的每個Li原子最多能吸附3個氫氣分子,因此我們在考慮各硅烯結構的氫氣吸附構型時,在每個Li原子附近擺放3個氫氣分子,擺放的初始位置和方向與Li0.11Si0.89硅烯中的氫氣分子類似,并且盡量使相鄰Li原子吸附的氫氣分子之間保持一定距離.經過多次氫氣分子的位置、方向調整和結構優化,最終以未被排斥遠離Li原子的氫氣分子數量作為該硅烯體系的最大儲氫數量.最終各硅烯體系的最大儲氫后結構和氫存儲性能可分別見圖6和表1所列.從圖6和表1中可以看到,隨著Li組分的不斷增加,儲氫量也不斷上升,表明提高Li吸附組分能夠有效提升Li修飾硅烯的氫存儲能力.Li0.20Si0.80,Li0.33Si0.67,Li0.43Si0.57和Li0.50Si0.50硅烯結構分別最多能夠吸附6個、8個、14個和18個氫氣分子,計算獲得的最大質量儲氫密度分別達到4.82 wt%,6.00 wt%,9.58 wt%和11.46 wt%,相應的平均吸附能分別為0.47 eV/H2,0.54 eV/H2,0.41 eV/H2和0.34 eV/H2.從表1中還可以看到,在計算中單獨使用GGA會嚴重低估氫氣分子的平均吸附能,而使用范德瓦耳斯修正能有效改善該問題,這與前人的報道相符合[8,14,22].最終可以發現,Li0.33Si0.67,Li0.43Si0.57和Li0.50Si0.50硅烯結構的最大質量儲氫密度和平均吸附能都達到了DOE制定的儲氫標準,能夠作為氫存儲材料使用,具有潛在的應用前景.
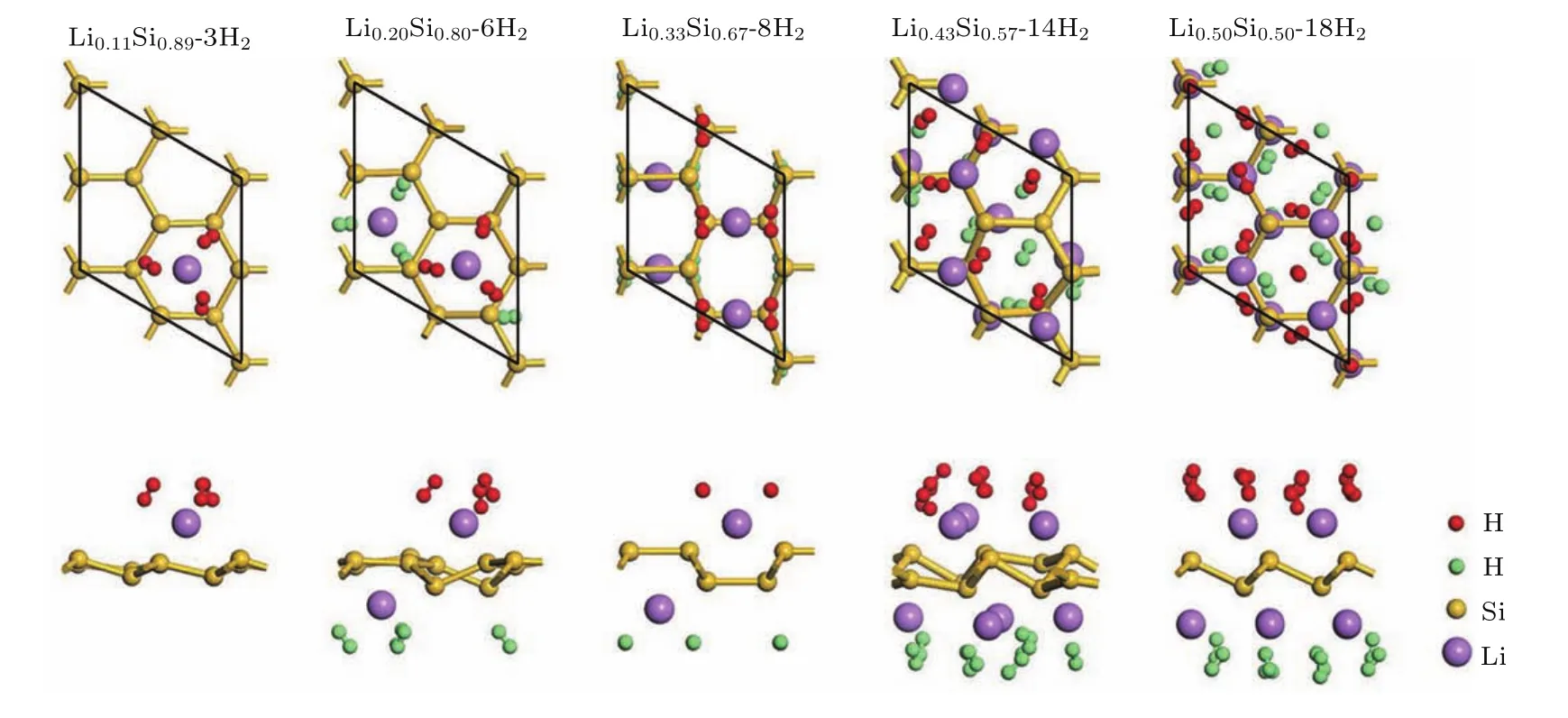
圖6 不同Li組分下硅烯體系的最大儲氫結構俯視和側視圖Fig.6.Top(upper panels)and side(bottom panels)views of geometry structures of LixSi1?xwith the maximum hydrogen storage.

表1 各硅烯體系的氫存儲性能Table 1.The hydrogen storage properties of LixSi1?x.
3.3.2 儲氫機理分析
為了探究Li修飾硅烯結構的氫吸附機理,本文以儲氫量最大的Li0.50Si0.50-18H2硅烯結構為例,對Mulliken電荷布居、差分電荷密度和態密度進行了分析.從Mulliken電荷布居可知,隨著Li0.50Si0.50硅烯不斷吸附氫氣分子,Li原子不斷失去電子,每個Li原子失去電子的數量從0.77個電子單調增加到1.15個.這一失去電子的單調過程會使電荷轉移形成的局域電場(Li正離子和Si負離子)不斷加強,并通過極化作用吸附更多的氫氣,這在差分電荷密度圖中可以清楚地看到(圖7(a)).黑色虛圓內的氫氣分子靠近Li原子一側的電荷密度增加(橙色),而遠離一側的電荷密度減少(藍色),從而說明該氫氣分子在局域強電場作用下被極化而形成了局部電偶極矩.因此,Li,Si離子同極化氫氣分子之間的靜電相互作用是Li修飾硅烯吸附氫氣的原因之一.
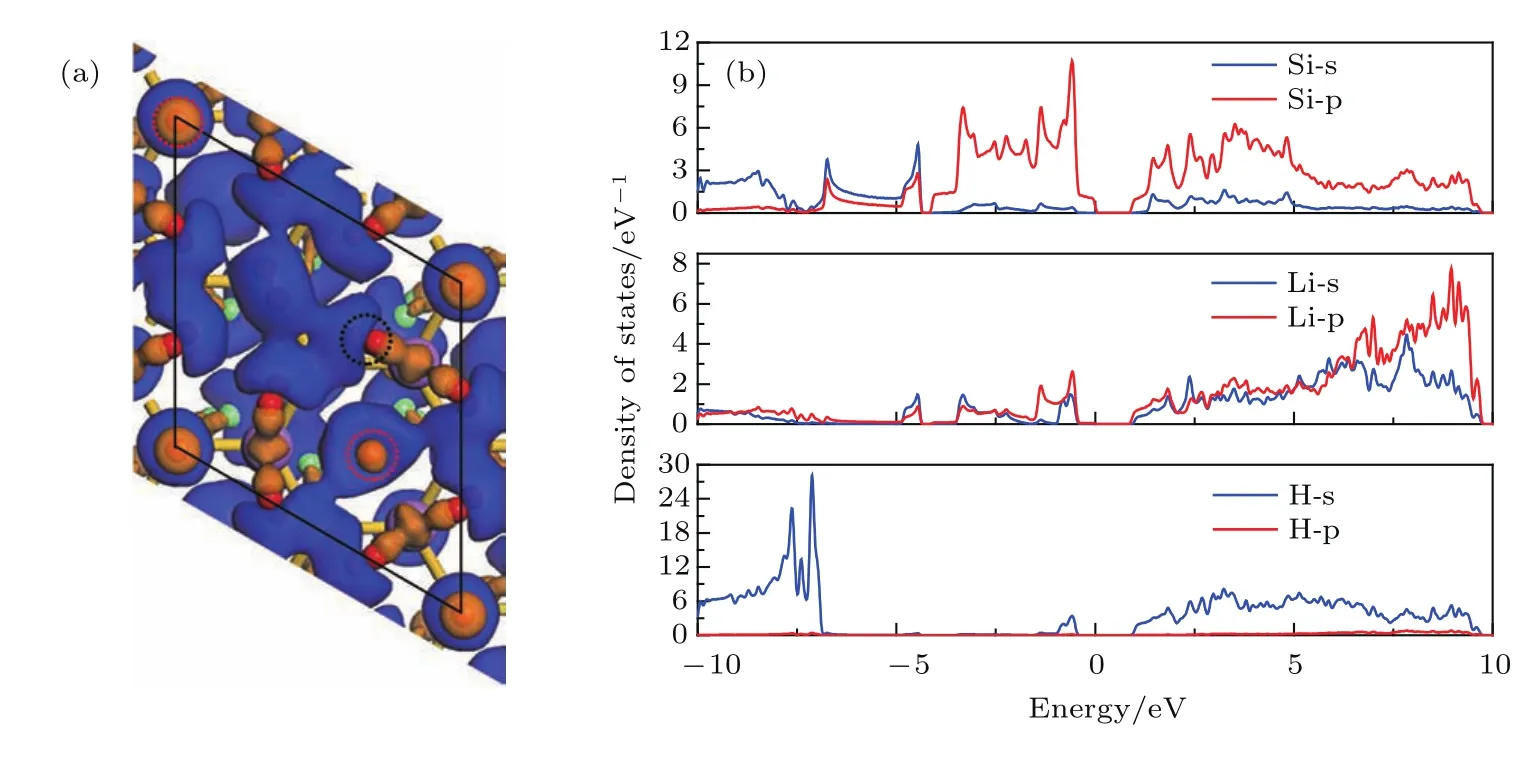
圖7 Li0.50Si0.50-18H2硅烯結構的(a)差分電荷密度圖,(b)態密度圖Fig.7.Diagrams of(a)charge density differences and(b)density of states of Li0.50Si0.50-18H2.
此外,從圖7(b)的態密度圖中可以看到,H的s和p軌道同Li,Si的s和p軌道存在不同程度的雜化(?0.95—?0.44 eV以及0.98—9.65 eV),說明軌道雜化作用在Li修飾硅烯氫存儲過程中也發揮了重要作用.結合Mulliken電荷布居的分析,發現各H原子從Li原子處獲得了數量不等的電子,說明軌道雜化作用加強了Li正離子、Si負離子和極化氫氣分子之間的靜電作用.值得注意的是,存在兩類氫氣分子(圖7(a)中紅色虛圓內)周圍的電荷密度增減情況與其他氫氣分子不同,且其吸附距離相較于其他各氫氣分子較大(2.37 ?和2.75 ?).本文嘗試在結構優化前將這兩類氫氣分子遠離硅烯表面,但在結構優化后這兩類氫氣分子仍然被吸附回硅烯表面,且其態密度圖和其他氫氣分子類似,因此我們認為上述兩類氫氣分子也被Li修飾硅烯所吸附,其吸附機理主要為軌道雜化作用.總體來看,氫氣分子的氫鍵鍵長在吸附后略有增長(由0.748 ?變為0.766 ?),表明靜電相互作用和軌道雜化作用只是促進了Li修飾硅烯對氫氣的吸附,并未將氫氣分子解離而破壞,這對于氫氣分子在Li修飾硅烯中的存儲和釋放非常重要.
4 結 論
借助第一性原理計算方法,對不同Li吸附組分下硅烯的穩定性、氫存儲性能和機理進行了系統性的研究.結果發現,在Li吸附組分達到0.50而飽和前,各硅烯體系中Li原子的平均結合能在大于其內聚能的基礎上,隨Li組分的增加而呈現增長趨勢,表明在一定范圍內提高Li修飾硅烯中的Li組分不會影響硅烯體系的穩定性,進一步說明本文研究不同Li組分下硅烯的氫存儲性能具有可行性.在確保穩定性的基礎上,通過依次添加氫氣分子,發現硅烯體系的最大氫存儲量會隨著Li組分從0.11到0.50的增加而增大,并且當Li原子吸附達到飽和時達到最大,相應的最大質量儲氫密度為11.46 wt%,平均吸附能為0.34 eV/H2,達到美國能源部制定的儲氫標準,表明提高Li吸附組分至其飽和在理論上能有效提高硅烯的氫存儲性能,使Li修飾硅烯成為良好的氫存儲材料.同時發現,Li,Si離子與極化氫氣分子之間的靜電相互作用和H,Li,Si的s和p軌道之間的雜化作用是Li修飾硅烯可具備較大儲氫能力的關鍵.上述研究結果揭示了Li修飾硅烯的儲氫機理,并為充分挖掘Li修飾硅烯的氫存儲性能和硅烯在未來氫存儲領域的應用提供了理論指導.
[1]Cheng J Y,Chan M K Y,Lilley C M 2016Appl.Phys.Lett.109 133111
[2]Zhou J Q,Bournel A,Wang Y,Lin X Y,Zhang Y,Zhao W S 2017Appl.Phys.Lett.111 182408
[3]Yang S,Cheng P,Chen L,Wu K H 2017Acta Phys.Sin.66 216805(in Chinese)[楊碩,程鵬,陳嵐,吳克輝2017物理學報66 216805]
[4]Hussain T,Kaewmaraya T,Chakraborty S,Ahuja R 2016J.Phys.Chem.C120 25256
[5]Li C,Yang S X,Li S S,Xia J B,Li J B 2013J.Phys.Chem.C117 483
[6]Li F,Zhang C W,Ji W X,Zhao M W 2015Phys.Status Solidi B252 2072
[7]Zhao J J,Liu H S,Yu Z M,Quhe R G,Zhou S,Wang Y Y,Liu C C,Zhong H X,Han N N,Lu J,Yao Y G,Wu K H 2016Prog.Mater.Sci.83 24
[8]Hussain T,Chakraborty S,De Sarkar A,Johansson B,Ahuja R 2014Appl.Phys.Lett.105 123903
[9]Wang Y S,Zheng R,Gao H Y,Zhang J,Xu B,Sun Q,Jia Y 2014Int.J.Hydrogen Energy39 14027
[10]Wang J,Li J B,Li S S,Liu Y 2013J.Appl.Phys.114 124309
[11]Ariharan A,Viswanathan B,Nandhakumar V 2017Graphene6 41
[12]Lochan R C,Head Gordon M 2006Phys.Chem.Chem.Phys.8 1357
[13]Song E H,Yoo S H,Kim J J,Lai S W,Jiang Q,Cho S O 2014Phys.Chem.Chem.Phys.16 23985
[14]Li F,Zhang C W,Luan H X,Wang P J 2013J.Nanopart.Res.15 1972
[15]Molle A,Grazianetti C,Cinquanta E 2016ECS Trans.75 703
[16]Zhong S Y,Ning F H,Rao F Y,Lei X L,Wu M S,Zhou L 2016Int.J.Mod.Phys.B30 1650176
[17]Hussain T,Kaewmaraya T,Chakraborty S,Ahuja R 2013Phys.Chem.Chem.Phys.15 18900
[18]Zhou C Y,Szpunar J A 2016ACS Appl.Mater.Interfaces8 25933
[19]Ma L,Zhang J M,Xu K W,Ji V 2015Physica E66 40
[20]Fair K M,Cui X Y,Li L,Shieh C C,Zheng R K,Liu Z W,Delley B,Ford M J,Ringer S P,Stampf lC 2013Phys.Rev.B87 014102
[21]Wang Y S,Li M,Wang F,Sun Q,Jia Y 2012Phys.Lett.A376 631
[22]Hussain T,Chakraborty S,Ahuja R 2013ChemPhys-Chem14 3463
[23]Delley B 2000J.Chem.Phys.113 7756
[24]Perdew J P,Chevary J A,Vosko S H,Jackson K A,Pederson M R,Singh D J,Fiolhais C 1993Phys.Rev.B48 4978
[25]Perdew J P,Wang Y 1992Phys.Rev.B45 13244
[26]Grimme S 2006J.Comput.Chem.27 1787
[27]Chadi D J 1977Phys.Rev.B16 1746
[28]Huang Y P,Yuan J M,Guo G,Mao Y L 2015Acta Phys.Sin.64 013101(in Chinese)[黃艷平,袁健美,郭剛,毛宇亮2015物理學報64 013101]
[29]Tritsaris G A,Kaxiras E,Meng S,Wang E G 2013Nano Lett.13 2258
[30]Liu C S,Zeng Z 2010Appl.Phys.Lett.96 123101
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
