Zn/Co-HMS催化氧化異戊醇生成異戊酸
2018-06-20 06:22:58胡乃寧趙彬俠劉林學關文斌韓銳暄吳笑江賀貝貝張小里
西北大學學報(自然科學版) 2018年3期
關鍵詞:催化劑
胡乃寧,趙彬俠,劉林學,關文斌,韓銳暄,吳笑江,岳 能,賀貝貝,張小里
(西北大學 化工學院, 陜西 西安 710069)
異戊酸是生產香料異戊酸酯的重要原料,存在于纈草油、酒花油中[1]。傳統工業生產常采用相轉移催化法和分步氧化法的方式制備異戊酸,但這兩種方法均需使用重金屬鹽,會對環境造成污染[2-3]。因此,選擇綠色醇氧化合成途徑具有重要意義。
以O2為氧化劑在催化劑作用下進行醇氧化具有原子利用率高,對環境無害的優點。Pd系、Ru系等均相催化劑用于醇類氧化已有報道,但此過程需在有機溶劑及高壓條件下進行,對生產條件要求較高,同時均相催化劑存在難分離、回收等缺點[4]。近年來,Au/TiO2[5],Pd/SBA-16[6],Au/SBA-15[7],Ag-HMS[8],PdAu/Zn-Al LDHs/GO[9]等用于醇類氧化的非均相催化劑相繼被報道,但此類催化劑的研究主要集中在貴重金屬上,增加了生產成本。因此,研發出一種經濟、高效的非均相催化劑對實際生產具有重要意義。
六方介孔硅分子篩(HMS)作為非均相反應催化劑的載體具有合成條件溫和、比表面積大、熱穩定性高等優點[10],白永燕等[11]綜述了近年HMS的改性方法,認為向HMS骨架中引入金屬,可大大提高HMS的催化性能。如Bhoware S S等[10]向HMS骨架中摻雜Co用于乙苯的氧化反應,Wang G J等[12]向HMS骨架中引入Zn用于噻吩的氧化反應, Saladino M L等[13]向HMS骨架中引入Ce用于乙醇部分氧化制乙醛中。另外,文獻[14-15]用Ru和Fe改性HMS用于醇的氧化反應中取得較好反應效果,文獻[16]中引入了Zn作為HMS改性劑,發現Zn能改變分子篩表面酸性并提高了產物選擇性。由此可見,通過向分子篩骨架中引入金屬(Co,Ce)與金屬直接改性(Zn,Fe等)可改善分子篩的催化性能。因此,本文采用Zn為HMS金屬改性劑并向其骨架中引入Co,制備了較為高效,綠色和經濟的非均相金屬改性HMS催化劑,并著重研究了Co和不同Zn源對異戊醇無溶劑催化氧化制備異戊酸的影響。
1 實 驗
1.1 試 劑
十二胺(DDA),正硅酸乙酯(TEOS),硝酸鋅[Zn(NO3)2·6H2O],硝酸鈷[Co(NO3)2·6H2O],乙酸鋅[Zn(CH3COO)2·2H2O],硫酸鋅[ZnSO4·7H2O],氯化鋅[ZnCl2],無水乙醇,異戊醇(以上均為分析純)。
1.2 催化劑制備
HMS的制備:以TEOS為硅源,DDA為模板劑,將3.453g DDA溶于30mL乙醇和水的混合物中攪拌30min,再將16 mL TEOS和無水乙醇混合后加入到攪拌后的混合物中,45℃晶化室溫老化后,再經離心、洗滌、干燥、550℃焙燒得到HMS。
Co-HMS(100)的制備:將DDA和0.208 1g硝酸鈷溶于乙醇和水的混合物中,攪拌30min得到A液,將正硅酸乙酯與乙醇混合后滴加入A液中,在45℃晶化后再經干燥、焙燒得到摻雜Co的HMS,記為Co-HMS(100)(Si/Co物質的量比為100∶1)。
Zn/Co-HMS(100)的制備:將一定量的Co-HMS(100)分子篩溶解在硝酸鋅溶液中浸漬12h,干燥后,再于550℃下焙燒4h即得到以硝酸鋅為鋅源的負載型催化劑(ZnO負載量為3%,質量分數),記為Zn/Co-HMS(100)-N。不同鋅源催化劑的制備過程同上,以乙酸鋅、硫酸鋅、氯化鋅為鋅源制備的催化劑分別記為Zn/Co-HMS(100)-Ac,Zn/Co-HMS(100)-S,Zn/Co-HMS(100)-Cl。
1.3 催化劑表征
催化劑物相分析使用的XRD衍射儀為日本島津公司生產的XRD-6100X,掃描速率10°/min,掃描范圍2°~80°。N2吸附脫附比表面積與孔隙度分析儀為康塔AUT0S0RB-1C型全自動物理化學吸附分析儀,300℃預處理3h后進行分析測定。FT-IR分析儀為Nicolet 6700型紅外光譜分析儀,波數測試范圍為400cm-1~4 000cm-1。TG分析在TG209F3型熱重分析儀上進行,升溫速率為10℃/min,溫度測試范圍為35℃~800℃。TPD分析儀為美國康塔公司的CBT-1化學吸附儀,升溫速率為10℃/min,掃描范圍50℃~800℃。催化劑表面形貌分析使用的是日立TM3000電子顯微鏡。
1.4 催化劑性能評價
異戊醇催化氧化反應如下:將15mL異戊醇、一定量催化劑加入裝有磁力攪拌與冷凝回流的三口燒瓶,以25mL/min的速率以鼓泡方式通入氧氣,恒溫下攪拌(600r/min)反應一定時間。產物在GC900A型氣相色譜儀上進行檢測分析(SE-30毛細管,FID檢測,柱箱溫度110℃,汽化及氫火焰溫度均為200℃)。
2 結果與討論
2.1 催化劑結構
2.1.1 XRD表征 圖 1為催化劑的XRD譜圖,從圖1(a)可以看出催化劑HMS,Co-HMS(100)及4種Zn源催化劑均在2θ=2°~3°出現HMS的(100)特征衍射峰,保留了HMS的介孔結構[14, 17-18]。但是,從圖1(a)中發現摻雜了Co之后HMS的(100)特征衍射峰從2θ=2.81°移至2.37°,這可能因為金屬Co(dCo2+=74 pm,dCo3+=63 pm)的離子半徑大于Si(dSi4+=42 pm)的離子半徑,當Co部分取代Si后使材料的結構發生了變化,說明Co已成功進入到HMS骨架中。此外,4種Zn源催化劑的(100)特征衍射峰強度進一步降低,可能因為ZnO粒子進入到催化劑孔道內引起[19]。從圖1(a),(b)中可以看出,負載后的催化劑未出現其他新峰,可能因為 Zn負載量少,且均勻分散于Co-HMS(100)表面及孔道中,因而在XRD中未出現衍射峰。
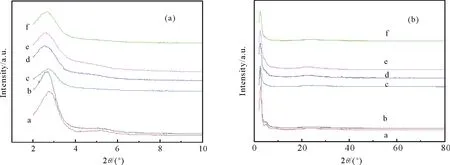
a HMS; b Co-HMS(100); c Zn/Co-HMS(100)-N; d Zn/Co-HMS(100)-Cl; e Zn/Co-HMS(100)-Ac; f Zn/Co-HMS(100)-S 圖1 不同催化劑低角度(a)和高角度(b)XRD譜圖Fig.1 XRD patterns of different samples (a) Low angle; (b) Wide angle
2.1.2 FT-IR表征 圖2為催化劑的FT-IR譜圖,可以看出摻雜Co和負載Zn后,催化劑的特征吸收振動峰基本不變,說明Co和Zn對HMS的結構未造成破壞,這與XRD得到的結論一致。其中,位于3 464cm-1和1 633cm-1的峰分別為載體上Si—OH伸縮振動吸收峰和吸附水—OH的彎曲振動吸收峰。位于1 085cm-1的峰為HMS中Si—O—Si的非對稱伸縮振動吸收峰,可觀察到吸收峰強度在摻雜Co之后明顯下降。這可能是由于Co進入HMS骨架引起。位于802cm-1,468cm-1的峰分別對應Si—O—Si對稱伸縮振動和彎曲振動峰[2, 21],位于966cm-1處的峰歸屬為HMS表面Si—OH的吸收峰。對比4種Zn源催化劑和Co-HMS(100)的紅外吸收峰,峰形相似且未發現新峰,說明Zn均勻分布在載體表面,未出現團聚。
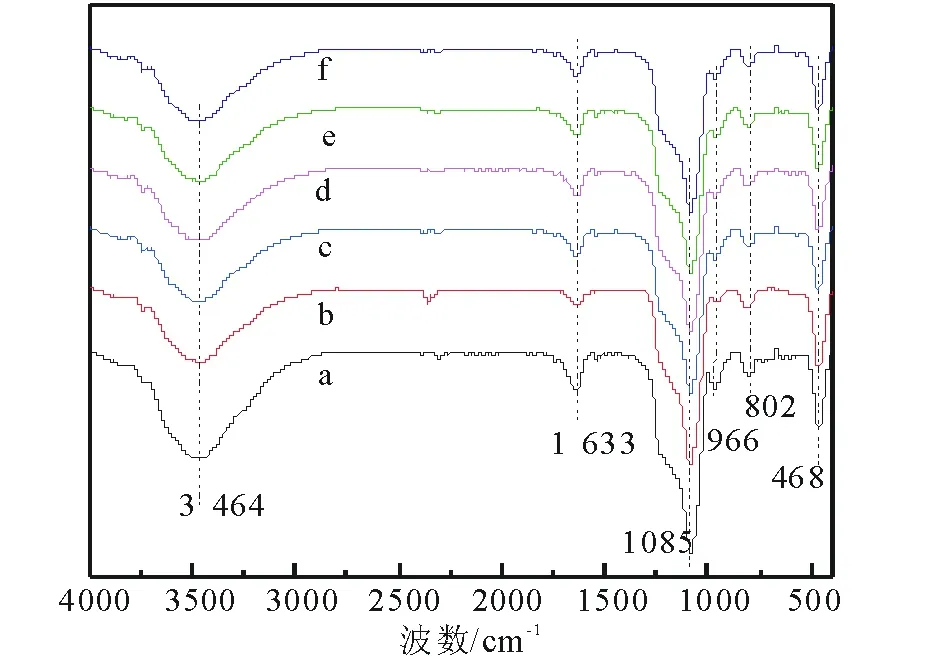
a HMS; b Co-HMS(100); c Zn/Co-HMS(100)-N; d Zn/Co-HMS(100)-Cl; e Zn/Co-HMS(100)-Ac; f Zn/Co-HMS(100)-S 圖2 不同催化劑的FT-IR譜圖Fig.2 The FT-IR spectrum of different catalysts
2.1.3 TG分析 對以硝酸鋅[Zn(NO3)2·6H2O],乙酸鋅[Zn(CH3COO)2·2H2O],硫酸鋅[ZnSO4·7H2O],氯化鋅[ZnCl2]為Zn源的催化劑未焙燒樣品做了TG-DTG分析,如圖3所示。
圖3中4種Zn源催化劑的焙燒溫度為550℃,均在50℃~100℃出現失重峰,此失重峰為樣品脫除吸附水引起。其中,(a)在250℃的失重峰為ZnCl2溶于水后形成的配酸H{ZnCl2(OH)}分解所致,當溫度升高至800℃未有失重峰出現,表明以ZnCl2為Zn源時,在焙燒溫度下是以ZnCl2的形式存在于載體上[22];(b)中在200℃~300℃的失重峰為Zn(CH3COO)2·2H2O中結合水分解所致,430℃左右的失重峰為乙酸鋅分解為ZnO,H2O和CO2所致,這表明以Zn(CH3COO)2·2H2O為Zn源時,在焙燒溫度下是以ZnO的形式存在于載體上[23]。(c)中500℃~600℃為ZnSO4·7H2O中結合水的失重峰,740℃附近為硫酸鋅分解失重峰,這表明以ZnSO4·7H2O為Zn源時,在焙燒溫度下是以ZnSO4的形式存在于載體上[24]。(d)在100℃~200℃的失重峰為Zn(NO3)2·6H2O結合水和硝酸鋅的分解峰,說明在焙燒之后是以ZnO的形式存在于載體上的[25]。
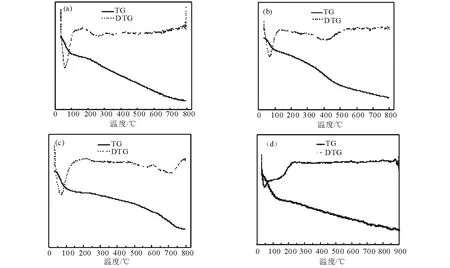
(a) Zn/Co-HMS(100)-Cl; (b) Zn/Co-HMS(100)-Ac; (c) Zn/Co-HMS(100)-S; (d) Zn/Co-HMS(100)-N 圖3 不同催化劑的TG-DTG圖譜Fig.3 TG-DTG spectrum of different catalysts
2.1.4 NH3-TPD酸性分析 圖4是催化劑的NH3-TPD圖譜,可知所有催化劑均在100℃~300℃和400℃~750℃出現了NH3脫附峰,分別對應弱酸位(L酸)和強酸位(B酸)[26]。對比a和b可知,Co-HMS(100)的弱酸含量幾乎不變而強酸含量略有增加,對比c,d和b可知Zn/Co-HMS(100)-N和Zn/Co-HMS(100)-Ac的弱酸含量明顯減小,而Zn/Co-HMS(100)-N的強酸含量略有增加,Zn/Co-HMS(100)-Ac的強酸含量略有減少。對比e,f和b發現,Zn/Co-HMS(100)-S和Zn/Co-HMS(100)-Cl的弱酸含量和強酸含量都大大增加。結合圖3可以得出:550℃焙燒后,Zn以ZnO形式存在的催化劑Zn/Co-HMS(100)-N與Zn/Co-HMS(100)-Ac的NH3-TPD圖譜曲線相似, Zn/Co-HMS(100)-N和Zn/Co-HMS(100)-Ac的弱酸含量相對Co-HMS(100)的弱酸含量減少,而Zn以ZnSO4和ZnCl2形式存在的催化劑Zn/Co-HMS(100)-S與Zn/Co-HMS(100)-Cl的弱酸含量較Co-HMS(100)的弱酸含量多。
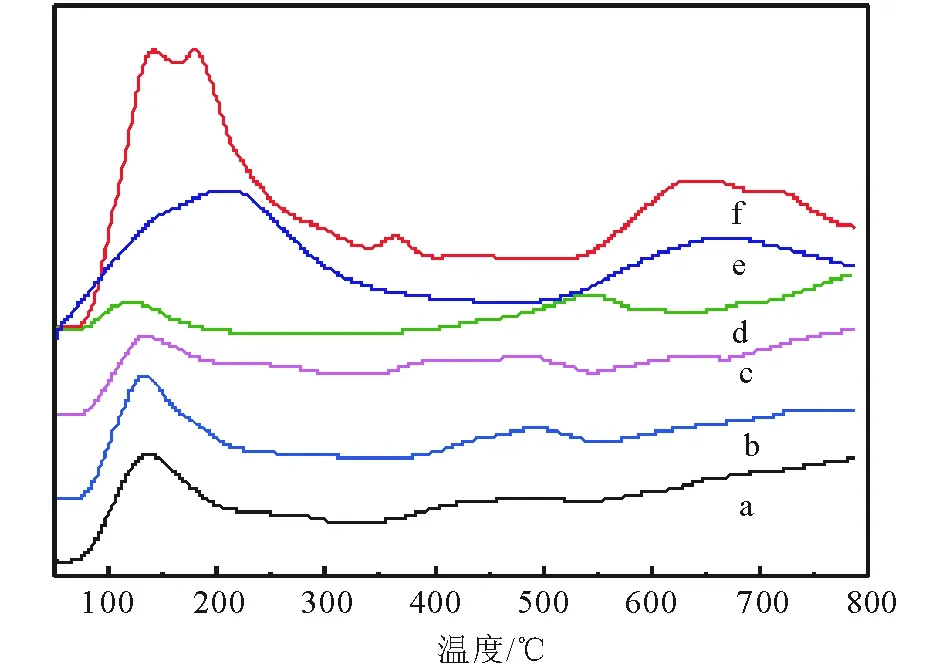
a HMS; b Co-HMS(100); c Zn/Co-HMS(100)-N; d Zn/Co-HMS(100)-Ac; e Zn/Co-HMS(100)-S; f Zn/Co-HMS(100)-Cl 圖4 不同催化劑的NH3-TPD圖譜Fig.4 NH3-TPD spectrum of different catalysts
2.1.5 N2吸附脫附 圖5是催化劑的N2吸附脫附等溫線圖譜。可以看出,所有樣品的吸附附脫等溫線均屬于IV型等溫線,且等溫線形狀相似,說明Co和Zn未對HMS的結構造成破壞,這與XRD及FT-IR得到的結論一致。圖6是催化劑的孔徑分布圖。可以看出,Co-HMS(100)的孔徑分布曲線右移,孔徑增大;Zn/Co-HMS(100)-N 和Zn/Co-HMS(100)-Ac的孔徑分布曲線相似。這可能是因為這兩種催化劑焙燒后Zn的存在形式都是ZnO;Zn/Co-HMS(100)-S的孔徑分布曲線左移,孔徑減小,這可能是因為ZnSO4分子較大,進入催化劑孔道后占據較大空間所致[27]; Zn/Co-HMS(100)-Cl孔徑分布曲線右移,這可能是因為ZnCl2堵塞小孔,小孔數量減小所致。表1為催化劑的結構參數,可以看出平均孔徑的變化與孔徑分布曲線的變化一致,催化劑的比表面積和總孔體積均隨著Co摻雜和不同Zn源負載而下降。
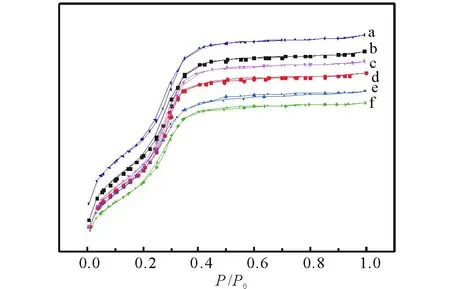
圖5 不同催化劑N2吸附脫附圖譜Fig.5 N2 adsorption-desorption isotherms of different catalysts
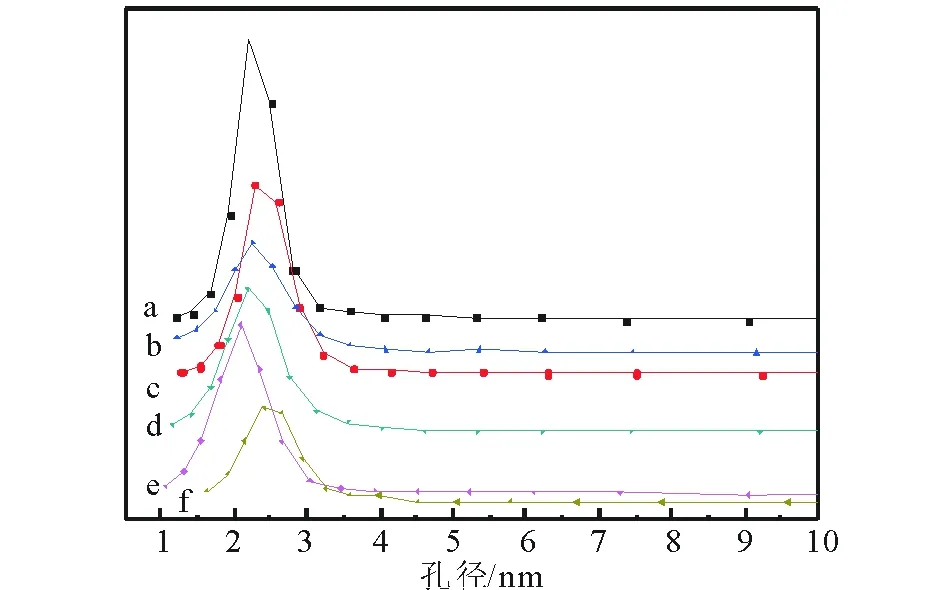
a HMS; b Co-HMS (100); c Zn/Co-HMS (100)-N; d Zn/Co-HMS (100)-Ac; e Zn/Co-HMS (100)-S; f Zn/Co-HMS (100)-Cl 圖6 不同催化劑孔徑分布圖譜Fig.6 The Pore size distribution of different catalysts
2.1.6 SEM形貌分析 為了探究催化劑的形貌對催化劑性能的影響,對4種Zn源催化劑做了SEM表征,如圖 7所示。可以看出,催化劑Zn/Co-HMS(100)-N,Zn/Co-HMS(100)-Ac,Zn/Co-HMS(100)-S的SEM圖相似,催化劑粒徑相近,但Zn/Co-HMS(100)-N粒度較均勻。Zn/Co-HMS(100)-Cl與其他3種催化劑相比, 其粒徑較大,面較粗糙,顆粒團聚現象較明顯。在催化反應中,較小的催化劑粒徑有利于催化劑積碳前驅體擴散[28],從而避免了催化劑積碳和失活。除此之外,較小的催化劑粒徑也有利于反應物與產物傳遞,促進反應進行。
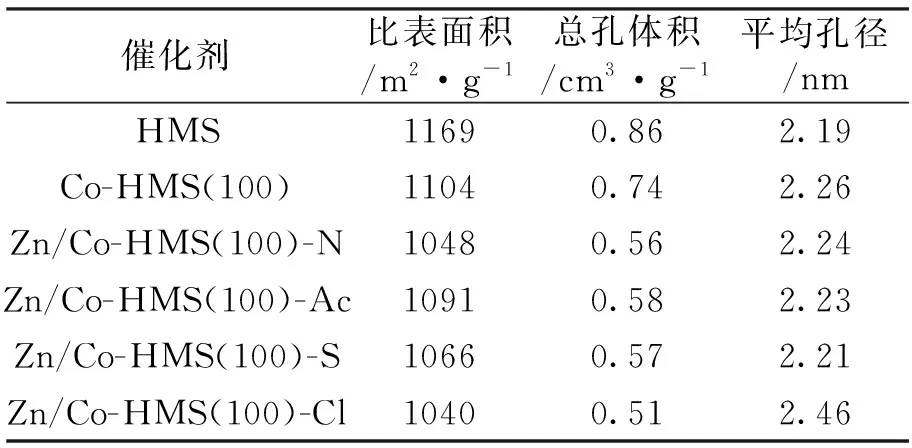
表1 不同催化劑的結構參數Tab.1 The structural parameters of different catalysts
表

a Zn/Co-HMS (100)-N; b Zn/Co-HMS (100)-Ac; c Zn/Co-HMS (100)-S; d Zn/Co-HMS (100)-Cl 圖7 不同催化劑的SEM圖Fig.7 SEM morphology micrograph of different catalysts
2.2 催化性能
表2中列出了不同催化劑在一定反應條件下的催化性能。其中,用X表示異戊醇的轉化率,S表示異戊酸的選擇性,Y表示異戊酸的收率(X=異戊醇的轉化量/異戊醇的起始量,S=產物異戊酸的量/異戊醇的轉化量,Y=產物異戊酸的量/異戊醇的起始量)。可以看出:Co-HMS(100)較HMS異戊醇的轉化率和異戊酸的收率與選擇性都有下降。Zn/Co-HMS(100)-N和Zn/Co-HMS(100)-Ac較Co-HMS(100)催化性能明顯增加,
其中Zn/Co-HMS(100)-N表現出較好的催化活性,異戊醇的轉化率、異戊酸的收率與選擇性達到52.2%,26.9%和51.5%。而Zn/Co-HMS(100)-S和Zn/Co-HMS(100)-Cl較 Co-HMS(100)催化活性下降。催化劑的活性與催化劑的表面性質及孔道結構有關。強酸(B酸)為重要的活性位點,大多存在于孔道內[29];而弱酸(L酸)是催化劑發生積碳的重要原因[30]。因此,較多的強酸、較少的弱酸及合適的孔道結構有利于催化反應發生。催化劑Zn/Co-HMS(100)-N和Zn/Co-HMS(100)-Ac弱酸含量少,減少了積碳發生,而Zn/Co-HMS(100)-N具有較合適的孔徑(圖6)和較小的粒徑(圖7)有利于反應物和產物傳遞,因此Zn/Co-HMS(100)-N催化效果優于Zn/Co-HMS(100)-Ac。催化劑Zn/Co-HMS(100)-S和Zn/Co-HMS(100)-Cl幾乎無催化活性,雖然催化劑Zn/Co-HMS(100)-Cl具有較大的孔徑(表 1)和較多的強酸含量(表4),但其弱酸含量多、粒徑大、催化劑團聚現象明顯(圖 7),導致催化劑積碳甚至失活,因此催化性能下降。除此之外,文中對反應的副產物進行了研究,如表 2所示, 催化劑Co-HMS異戊醛的選擇性為23.5%,較HMS和其他四種負載型催化劑高,而Co-HMS中幾乎不產生異戊酸異戊酯;催化劑Zn/Co-HMS(100)-N和 Zn/Co-HMS(100)-Ac異戊醛選擇性分為6.7%和4.5%,低于HMS,而Zn/Co-HMS(100)-N和 Zn/Co-HMS(100)-Ac異戊酸異戊酯的選擇性相對較高分別為13.6%和12.8%。Zn/Co-HMS(100)-S和Zn/Co-HMS(100)-Cl中幾乎無異戊醛與異戊酸異戊酯生成。
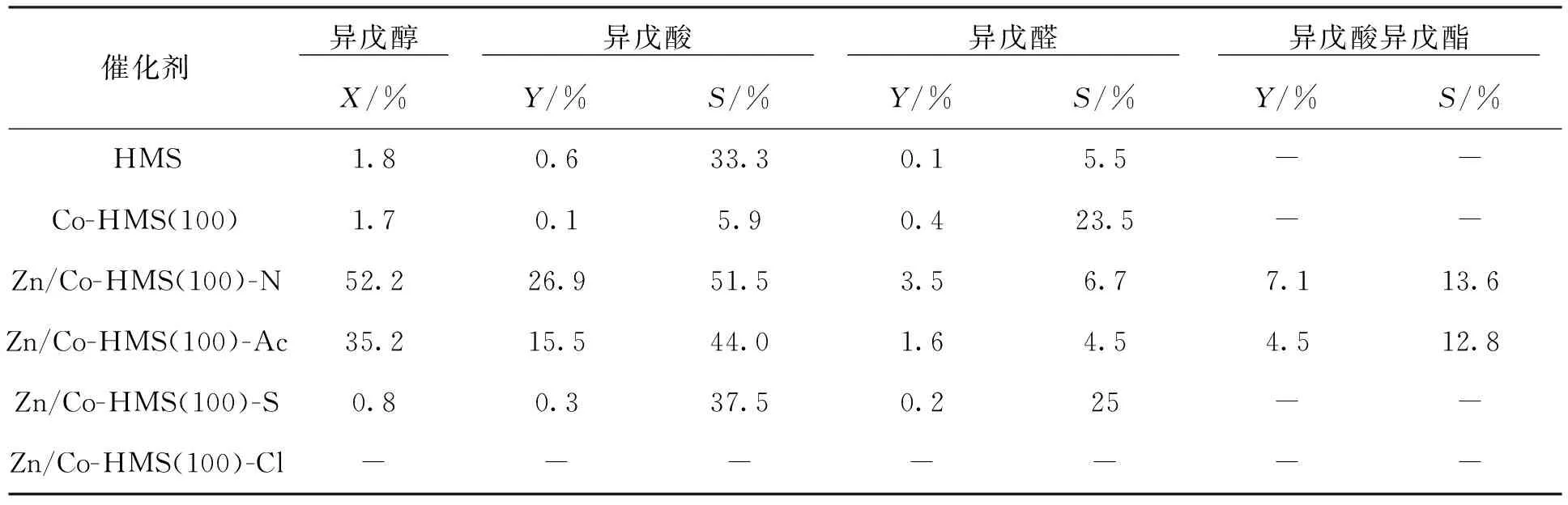
表2 不同催化劑的催化性能Tab.2 Catalytic performance of different catalysts
注:反應條件為溫度, 120℃; 異戊醇, 15mL; 催化劑, 0.5g;反應時間, 8h; O2, 25mL/min。
2.3 催化反應機理
為探究異戊醇催化氧化生成異戊酸的反應機理,做了以下5組實驗:a 向體系中加入0.5 g HMS 不通入O2;b向體系中加入0.5 g HMS并通入O2;c 向體系中加入0.5 g Co-HMS(100)并通入O2;d向體系中加入0.5g Zn/Co-HMS(100)-N并通入O2;e向體系中加入少量水,0.5g Zn/Co-HMS(100)-N并通入O2反應結果如表 3所示。
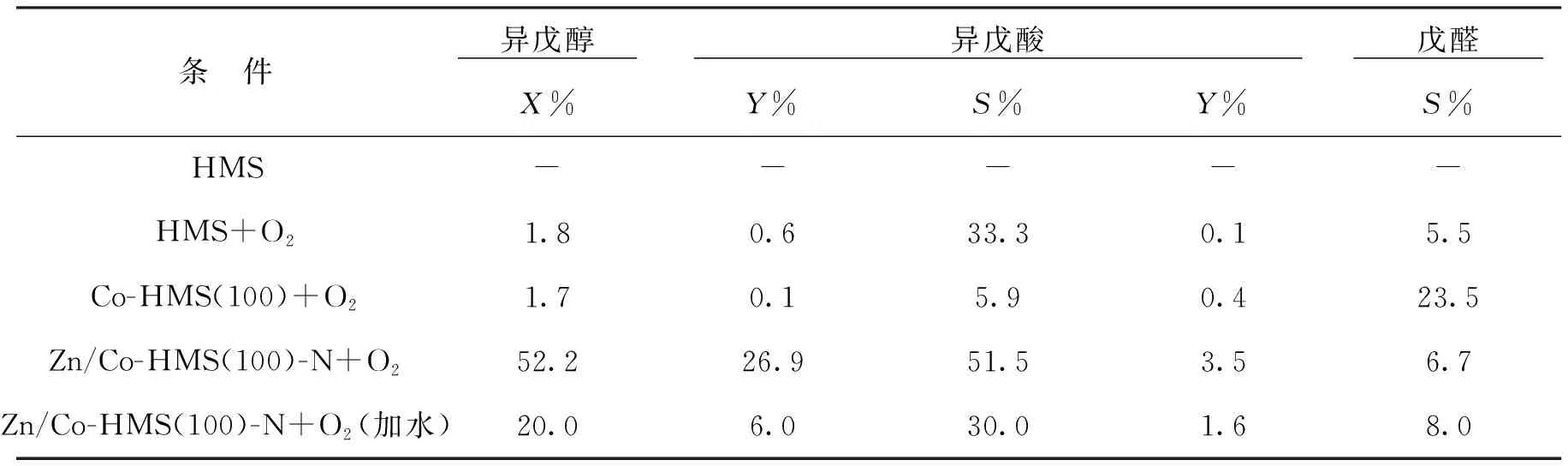
表3 不同反應條件下異戊醇催化氧化效果Tab.3 The catalytic oxidation results of isoamyl alcohol under different reaction conditions
反應條件: 溫度120℃; 異戊醇15mL; 催化劑0.5g;反應時間8h; O225mL/min。
可以看出,當反應體系中加入HMS不通入O2時,產物中未發現異戊酸和異戊醛;當反應體系中加入HMS并通入O2時,有少量異戊酸和異戊醛的產生;當反應體系中加入Co-HMS(100) 并通入O2時,發現產物中異戊醛的含量多于異戊酸的含量;反應體系中加入Zn/Co-HMS(100)-N 并通入O2時,發現異戊酸的含量較多且大于異戊醛的含量。綜上所述,在異戊醇生成異戊酸的反應中Co能促進異戊醛生成,Co和Zn協同能促進異戊酸生成,O2在異戊醇生成異戊酸的整個過程都發揮了重要作用。文獻[31-32]中研究發現有O2參與的羰基化合物制備中,氧氣活化是反應的關鍵部分,過渡態金屬如Cr,Co等具有未充滿的價層d軌道能使O2激活,成為活化態氧。異戊醛中羰基CO的π鍵較δ鍵弱,易受到周圍原子電子云的影響而斷裂[33-34],Zn外層電子為4s23d10電子軌道全充滿,電子云密度較大,會對CO的π鍵產生較大影響,使π鍵發生斷裂。異戊醇及異戊醛分子結構特點及催化劑特性符合自由基反應歷程,其可能的反應路線如圖8所。
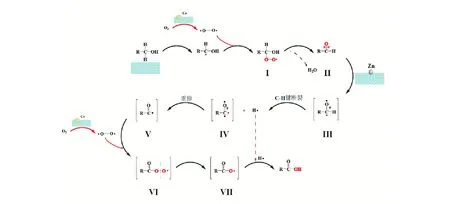
圖8 Zn/Co-HMS(100)-N催化氧化異戊醇生成異戊酸反應機理(R=CH3CH(CH3)CH2)Fig.8 Reaction mechanism of isoamyl alcohol to isovaleric acid under Zn/Co-HMS(100)-N
第一步:Co原子激發產生的過氧自由基·O—O·與活化后的異戊醇生成中間體Ⅰ,中間體Ⅰ脫去1分子水生成異戊醛(Ⅱ)。反應第二步:異戊醛羰基中π鍵斷裂生成中間體Ⅲ,Ⅲ中C—H易斷裂生成中間體Ⅳ及H·,Ⅳ不穩定重排后生成中間體Ⅴ,Ⅴ與過氧自由基生成過氧酸Ⅵ,Ⅵ中脫去一個O·后得到中間體Ⅶ,Ⅶ與Ⅲ中產生的H·生成最終的異戊酸,Ⅵ中脫去的O·與下一輪反應中產生的O·生成·O—O·參與到反應中去。
由于自由基反應過程中有水產生,因此水會抑制自由基反應進行。如表3所示,向反應體系中加入少量水對提出的機理進行驗證,可以看出添加水后,異戊醇的轉化率,異戊酸的收率與選擇性都減小,說明水抑制了反應進行,也佐證了該反應符合自由基機理。
3 結 論
本文研究了HMS,Co-HMS(100)及不同Zn源催化劑Zn/Co-HMS(100)-N,Zn/Co-HMS(100)-Ac,Zn/Co-HMS(100)-S和Zn/Co-HMS(100)-Cl用于異戊醇催化氧化制備異戊酸的反應效果。結果發現:不同Zn源催化劑均保留了載體的原有結構;焙燒后以ZnO存在的催化劑Zn/Co-HMS(100)-N。Zn/Co-HMS(100)-Ac具有較合適的比表面積,孔道結構及合適的酸含量及酸強度;焙燒后以ZnSO4和ZnCl2形式存在的催化劑Zn/Co-HMS(100)-S與Zn/Co-HMS(100)-Cl的弱酸含量增加。催化劑性能測試結果表明Zn/Co-HMS(100)-N較其他催化劑反應效果好,異戊醇的轉化率、異戊酸的收率與選擇性分別可以達到52.2%,26.9%和51.5%。而Zn/Co-HMS(100)-S和Zn/Co-HMS(100)-Cl幾乎無催化活性。以Zn/Co-HMS(100)-N為催化劑,對異戊醇催化氧化生成異戊酸的反應機理進行了初步探討,發現該反應符合自由基機理,催化劑的主要活性位點是Co與Zn,Co和Zn協同作用催化異戊醇生成異戊酸。
參考文獻:
[1] 何優選, 李善吉. 香料異戊酸芐酯的綠色合成工藝研究[J]. 廣州化工, 2010, 38(4): 96-97.
[2] WANG X, KAWANAMI H. Selective oxidation of alcohols to aldehydes and ketones over TiO2-supported gold nanoparticles in supercritical carbon dioxide with molecular oxygen[J]. Applied Catalysis A General, 2008, 349(1): 86-90.
[3] HADDADI H, KORANI E M, HAFSHEJANI S M, et al. Highly selective oxidation of sulfides to sulfones by H2O2catalyzed by porous capsules[J].Journal of Cluster Science, 2015, 26(6): 1-10.
[4] MA C Y, CHENG J, WANG H L, et al. Characteristics of Au/HMS catalysts for selective oxidation of benzyl alcohol to benzaldehyde[J]. Catalysis Today, 2010, 158(3-4): 246-251.
[5] ZHOU L, CHEN M, Wang Y, et al. Au/mesoporous-TiO2, as catalyst for the oxidation of alcohols to carboxylic acids with molecular oxygen in water[J]. Applied Catalysis A General, 2014, 475(1): 347-354.
[6] CHEN Y, LIM H, TANG Q, et al. Solvent-free aerobic oxidation of benzyl alcohol over Pd monometallic and Au-Pd bimetallic catalysts supported on SBA-16 mesoporous molecular sieves[J]. Applied Catalysis A General, 2010, 380(1-2): 55-65.
[7] MA C Y, DOU B J, LI J J, et al. Catalytic oxidation of benzyl alcohol on Au or Au-Pd nanoparticles confined in mesoporous silica[J]. Applied Catalysis B Environmental, 2009, 92(1): 202-208.
[8] JIA L, ZHANG S, GU F, et al. Highly selective gas-phase oxidation of benzyl alcohol to benzaldehyde over silver-containing hexagonal mesoporous silica[J]. Microporous & Mesoporous Materials, 2012, 149(1): 158-165.
[9] 馬帥, 陳鄭, 王家喜. 氧化石墨烯摻雜鋅鋁類水滑石負載鈀金催化劑的制備及催化性能[J]. 化工進展, 2017,36(11): 4087-4092.
[10] BHOWARE S S, SHYLESH S, KAMBLE K R, et al. Cobalt-containing hexagonal mesoporous molecular sieves (Co-HMS): Synthesis, characterization and catalytic activity in the oxidation reaction of ethylbenzene[J]. Journal of Molecular Catalysis A Chemical, 2006, 255(1): 123-130.
[11] 白永燕, 陳平. 介孔材料HMS的改性及在催化反應中的應用[J]. 現代化工, 2016(10): 54-58.
[12] WANG G J, ZENG N. Preparation and catalytic properties of Zn-HMS molecular sieves[J]. Advanced Materials Research, 2012, 560-561: 300-304.
[13] SALADINO M L, MARTINO D F C, KRALEVA E, et al. Effect of the cerium loading on the HMS structure. Preparation, characterization and catalytic properties[J]. Catalysis Communications, 2013, 36(36): 10-15.
[14] 紀紅兵, 錢宇, 王婷婷,等. 負載釕的HMS催化劑催化氧化醇[J]. 化工學報, 2006, 57(3): 577-581.
[15] 賈麗華, 張森, 宋賀,等. 鐵改性HMS催化氧化苯甲醇合成苯甲醛[J]. 化工學報, 2009, 60(9): 2210-2214.
[16] 劉維橋, 雷衛寧, 尚通明,等. Zn對HZSM-5分子篩催化劑物化及甲醇芳構化反應性能的影響[J]. 化工進展, 2011, 30(9): 1967-1971.
[17] ZhANG P, ZHOU Y, FAN M, et al. Catalytic performance of PdCl2/Cu-HMS: Influence of hydrophobicity and structure of molecular sieves[J]. Applied Surface Science, 2014, 295(6): 50-53.
[18] PARSAFARD N, PEYROVI M H, RASHIDZADEH M. n-Heptane isomerization on a new kind of micro/mesoporous catalyst: Pt supported on HZSM-5/HMS[J]. Microporous & Mesoporous Materials, 2014, 200: 190-198.
[19] MATE V R, SHIRAI M, RODE C V. Heterogeneous Co3O4, catalyst for selective oxidation of aqueous veratryl alcohol using molecular oxygen[J]. Catalysis Communications, 2013, 33(9): 66-69.
[20] SELVARAJ M, LEE T G. t-Butylation of toluene with t-butyl alcohol over mesoporous Zn-Al-MCM-41 molecular sieves[J]. Microporous & Mesoporous Materials, 2005, 85(1): 59-74.
[21] ZHANG N, LI G, CHENG Z, et al. Rhodamine B immobilized on hollow Au-HMS material for naked-eye detection of Hg2+in aqueous media.[J]. Journal of Hazardous Materials, 2012, 229-230(43): 404-410.
[22] BI Y, WANG Y, CHEN X, et al. Methanol aromatization over HZSM-5 catalysts modified with different zinc salts[J]. Chinese Journal of Catalysis, 2014, 35(10): 1740-1751.
[23] 趙新宇, 李春忠, 鄭柏存,等. 二水合醋酸鋅熱分解機理與動力學[J]. 華東理工大學學報, 1997,23(2):191-195.
[24] 李國強. 無機含氧酸及其鹽熱穩定性研究[J]. 西安建筑科技大學學報(自然科學版), 1997,29(1):69-73.
[25] 戴長文. 離子極化和硝酸鹽熱穩定性勢標度[J]. 鄭州大學學報(理學版), 1985,32(2):67-72.
[26] Jia A, Lou L L, Zhang C, et al. Selective oxidation of benzyl alcohol to benzaldehyde with hydrogen peroxide over alkali-treated ZSM-5 zeolite catalysts[J]. Journal of Molecular Catalysis A Chemical, 2009, 306(1-2):123-129.
[27] CANG R, LU B, LI X, et al. Iron-chloride ionic liquid immobilized on SBA-15 for solvent-free oxidation of benzyl alcohol to benzaldehyde with H2O2[J]. Chemical Engineering Science, 2015, 137: 268-275.
[28] RASOULI M, ATASHI H, MOHEBBI-KALHORI D, et al. Bifunctional Pt/Fe-ZSM-5 catalyst for xylene isomerization[J]. Journal of the Taiwan Institute of Chemical Engineers, 2017,78: 438-446.
[29] ZHAO Y X, BAMWENDA G R, GROTEN W A, et al. The chain mechanism in catalytic cracking: The kinetics of 2-methylpentane cracking[J]. Journal of Catalysis, 1993, 140(1): 243—261.
[30] BARZETTI T, SELLI E, MOSCOTTI D, et al. Pyridine and ammonia as probes for FTIR analysis of solid acid catalysts[J]. Journal of the Chemical Society Faraday Transactions, 1996, 92(8): 1401-1407.
[31] LIU P, DEGIRMENCI V, HENSEN E J M. Unraveling the synergy between gold nanoparticles and chromium-hydrotalcites in aerobic oxidation of alcohols[J]. Journal of Catalysis, 2014, 313(313): 80-91.
[32] BESSON M, GALLEZOT P. Selective oxidation of alcohols and aldehydes on metal catalysts[J]. Catalysis Today, 2000, 57(1): 127-141.
[33] 巨勇, 席嬋娟, 趙國輝. 有機合成化學與路線設計[M]. 北京: 清華大學出版社, 2007:116-121.
[34] 陳治明. 有機合成原理及路線設計[M]. 北京: 化學工業出版社, 2010:50-58.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
