基于精準(zhǔn)醫(yī)療的兩例基因突變患兒病例分析
2018-07-02 06:26:20姚獻(xiàn)花
中國全科醫(yī)學(xué) 2018年17期
姚獻(xiàn)花
在既往的康復(fù)工作中,對發(fā)育落后、運動障礙、行為異常、認(rèn)知及語言障礙等患兒的治療常選用傳統(tǒng)的康復(fù)治療方法,如按摩、推拿、針灸、語言認(rèn)知訓(xùn)練及現(xiàn)代康復(fù)訓(xùn)練的運動療法(PT)、作業(yè)療法(OT)等,部分患兒可取得較好的療效;但是,對先天因素所導(dǎo)致的發(fā)育異常患兒,常效果甚微。隨著分子醫(yī)學(xué)技術(shù)的發(fā)展及廣泛應(yīng)用于臨床,這些疑難病癥能夠給予明確的診斷,并對康復(fù)治療及預(yù)后的判斷給予正確、精準(zhǔn)的指導(dǎo)。本文通過對2例罕見病例的討論,目的是闡述基因檢測在康復(fù)醫(yī)學(xué)領(lǐng)域中的重要作用;其意義是建議醫(yī)生在臨床工作中不僅對患者的臨床癥狀、體征及其他實驗室檢測指標(biāo)要綜合分析,必要時結(jié)合基因檢測技術(shù)以明確診斷、指導(dǎo)治療及預(yù)后評估。
1 病例簡介
患兒1,男,1歲2個月,漢族,以發(fā)現(xiàn)“下肢不支持體質(zhì)量3個月”為代主訴于2015-05-06收入河南中醫(yī)藥大學(xué)第一附屬醫(yī)院。入院癥見:拱背坐,不會爬,不會獨站、獨走,立位雙下肢支持體質(zhì)量差,納食可,二便正常。個人史:患兒系G2P2,孕36+5周,順產(chǎn),出生體質(zhì)量2.8 kg,出生后哭聲可,出生后病理性黃疸,黃疸于1個月內(nèi)消退,混合喂養(yǎng),無易驚、易激惹病史。既往史:出生后有病理性黃疸病史,否認(rèn)藥物、食物過敏史,已按計劃進(jìn)行預(yù)防接種。發(fā)育史:3個月抬頭,8個月坐,8個月翻身。
體格檢查:一般體格檢查無異常,但患兒四肢及軀干部皮膚干燥粗糙如魚鱗,手足有大面積脫皮,面部皮膚細(xì)白。
專科檢查:追視追聽可,能笑出聲,會無意識發(fā)“ba”“o”音。會主動抓物,拇指內(nèi)收,可拇示指對捏;豎頭穩(wěn),會主動翻身;坐位拱背坐,坐位側(cè)方平衡(+);俯臥位肘支撐,短時手支持,抬胸,可抬頭90°;扶站時雙下肢可短時支持體質(zhì)量,足外翻,下肢稍外旋,屈髖,不會獨站,可短時扶欄站立;雙上肢肌張力不高,踝膝關(guān)節(jié)被動活動時稍抵抗,下肢肌張力高,股角110°,腘角130°,足背屈角60°,肌力降低,下肢肌力4+級。原始反射:擁抱反射(-),非對稱性緊張性頸反射(ATNR)(-),對稱性緊張性頸反射(STNR)(-),側(cè)彎反射(+),緊張性迷路反射(TLR)(-),手握持反射(-),足把握反射(+)。落傘反射(-),頸軟無抵抗,雙側(cè)膝腱反射亢進(jìn),雙側(cè)巴氏征(+),踝陣攣(-)。
輔助檢查:染色體檢查未見異常(2015-03-11外院檢查)。
入院診斷:(1)腦性癱瘓;(2)遺傳代謝病?(3)魚鱗病?
診療經(jīng)過:入院后給予綜合康復(fù)治療;并于2015-06-29檢查顱腦磁共振成像未見異常,2015-12-28遺傳代謝病篩查未見典型改變,生物素未見異常;給予生物素口服試驗性治療后患兒皮膚粗糙、雙手/足蛻皮現(xiàn)象較前好轉(zhuǎn),但仍不能站立與行走,下肢肌張力增高,膝腱反射亢進(jìn),踝陣攣(+),因治療時間較長,效果不明顯,建議到北京進(jìn)一步明確診斷,至北京大學(xué)第一附屬醫(yī)院檢查顱腦磁共振成像未見異常,血常規(guī)及生化未見異常,考慮診為:(1)神經(jīng)變性病;(2)線粒體病?(3)溶酶體病?建議做基因檢查,2016-03-15,由北京福佑龍惠遺傳病專科門診基因檢測公司經(jīng)基因檢測后確診為Sj?gren-Larsson綜合征。患兒基因檢測結(jié)果見表1、圖1。
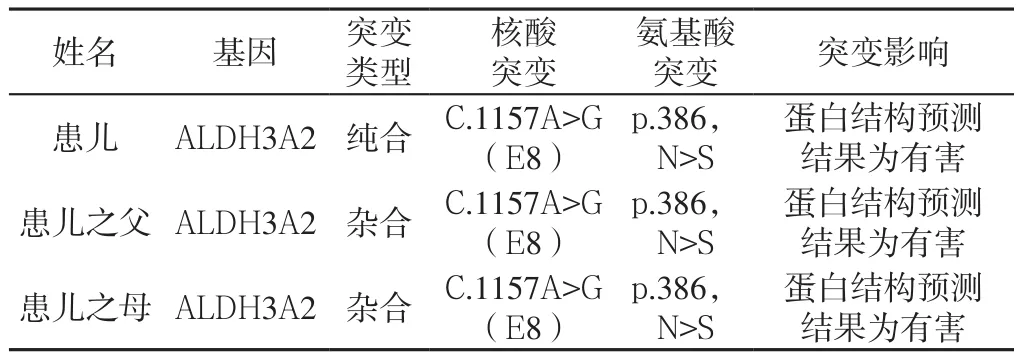
表1 患兒1及父母基因檢測結(jié)果Table 1 Genetic test results of case 1(14-month-old male child) of genic mutation and his parents
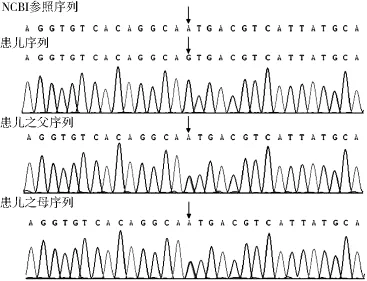
圖1 患兒1及其父母ALDH3A2基因測序圖Figure 1 ALDH3A2 genomic sequence of case 1(14-month-old male child) of genic mutation and his parents
患兒2,男,9個月,因“發(fā)現(xiàn)運動發(fā)育落后5月余”于2016-05-05入住河南中醫(yī)藥大學(xué)第一附屬醫(yī)院。現(xiàn)病史:5個月前(3月齡時)患兒豎頭不穩(wěn),不會翻身,當(dāng)時檢查顱腦磁共振成像提示蛛網(wǎng)膜下腔增寬,枕大池擴大,查體肋骨外翻,肌力低下,未予重視;4個月前至武漢某醫(yī)院考慮為戊二酸尿癥,檢查遺傳代謝尿篩查、肌電圖未見異常,心肝膽胰脾腎臟彩超未見異常。生化提示肝損傷,給予肌苷片、聯(lián)苯雙酯等保肝降酶藥物治療(1個月后復(fù)查降至正常),后至襄陽市某院診斷為中樞性協(xié)調(diào)障礙,給予綜合康復(fù)治療1個月余,患兒仍然豎頭不穩(wěn),不會翻身,姿勢異常,體質(zhì)量不增;后至北京兒童醫(yī)院復(fù)查磁共振成像結(jié)果顯示雙側(cè)豆?fàn)詈思拔矤詈宋s伴長T2信號,雙側(cè)腦室擴張,雙側(cè)腦室后部白質(zhì)T2W信號偏高,腦干聽覺誘發(fā)電位異常,喉鏡提示喉軟化癥,尿有機酸分析未見異常,血氨基酸提示多種氨基酸降低,丁二酰肉堿增高,提示短鏈脂肪酸代謝不暢,乳酸、血氨、銅藍(lán)蛋白、巨細(xì)胞病毒、EB病毒均陰性,診斷為:(1)發(fā)育倒退;(2)肝功異常;(3)先天性喉喘鳴;給予保肝治療,并行全套基因篩查,家屬為求進(jìn)一步治療遂至本院。入院癥見運動發(fā)育落后,豎頭不穩(wěn),不會翻身,不會坐,雙下肢不支持體質(zhì)量,易驚,喉間痰鳴,四肢姿勢不對稱,緊張狀態(tài)下四肢不隨意運動更明顯,納眠一般,二便正常。
個人史:患兒系G1P1,孕40周后行剖宮產(chǎn)(因家屬考慮妊娠期過長,要求剖宮產(chǎn)),出生體質(zhì)量3.5 kg,出生過程順利,無產(chǎn)傷及窒息史,哭聲可,新生兒期易驚顫,有哭鬧,黃疸持續(xù)約1個月。家族史:父母體健;否認(rèn)家族遺傳病病史。父母非近親結(jié)婚。體格檢查:意識清,精神可,追視可,追聽反應(yīng)差,可認(rèn)人,與人交流意識差,表情淡漠,反應(yīng)遲鈍,形體消瘦,面色無華,四肢活動少,未見通貫掌。頭圍45 cm,前囟1.5 cm×1.5 cm,平軟,體質(zhì)量7.5 kg。眼瞼無水腫,無下垂,雙側(cè)瞳孔等大等圓,對光反射靈活。氣管居中,雙肺聽診呼吸音清,未聞及干濕啰音。心率:110次/min,心音有力,律齊,未聞及病理性雜音。腹軟,無膨隆,肝脾肋下未觸及,腸鳴音正常。肋骨稍外翻,脊柱四肢無畸形,肛門及外生殖器無異常。
專科檢查:追視可,追聽反應(yīng)差。雙手拇指內(nèi)收,無主動抓物意識。豎頭不穩(wěn),不會翻身,不會坐,仰臥位四肢姿勢不對稱;側(cè)臥位呈角弓反張狀,俯臥位肘不支撐,抬頭15°~45°;立位下肢不支持體質(zhì)量;安靜狀態(tài)下四肢肌力、肌張力偏低;雙上肢肩肘關(guān)節(jié)松弛,活動無抵抗,內(nèi)收角60°~80°,腘角150°~180°,足背屈角30°,四肢肌力約3級。Vojta姿勢反射:拉起反射頭后墜,無主動抬頭意識;立位懸垂反射下肢伸展,雙足交叉;俯臥懸垂反射軀干呈倒“U”型。原始反射:擁抱反射伸展相,側(cè)彎反射(+),TLR(+);ANTR(+);STNR(-);手足把握反射(+);雙側(cè)膝腱反射可引出,踝陣攣(-),雙側(cè)巴氏征(+)。
住院期間完善檢查:血常規(guī):白細(xì)胞計數(shù)5.64×109/L,中性粒細(xì)胞分?jǐn)?shù)0.198,淋巴細(xì)胞分?jǐn)?shù)0.666;大便常規(guī)未見異常,傳染病四項未見異常,甲狀腺功能未見異常,優(yōu)生優(yōu)育十項未見異常。生化指標(biāo):血氨41 μmol/L,丙酮酸127 μmol/L,總膽固醇2.73 mmol/L,三酰甘油2.32 mmol/L,丙氨酸氨基轉(zhuǎn)移酶(ALT)49 U/L,天冬氨酸氨基轉(zhuǎn)移酶(AST)65 U/L,肌酸激酶206 U/L,肌酸激酶同工酶32 U/L;銅藍(lán)蛋白0.25 g/L,同型半胱氨酸5.90 μmol/L,乳酸2.10 mmol/L,葡萄糖4.39 mmol/L;25-羥維生素D 39.49 nmol/L。24 h腦電圖示:正常嬰兒腦電圖。基因檢測結(jié)果顯示(北京信諾佰世醫(yī)學(xué)檢驗所):線粒體DNA耗竭綜合征9型(腦肌病型)伴甲基丙二酸尿癥。基因測序提示SUCLG1基因發(fā)現(xiàn)2個雜合突變點,分別為c.961C>G(p.A321P)和c.713T>C(p.D238 G)(見表2、圖2~3),遺傳方式為常染色體隱性遺傳。其中c.961C>G雜合突變?yōu)橐褕蟮赖闹虏⊥蛔儯瑏碜阅赣H;c.713T>C雜合突變來自父親,目前未見文獻(xiàn)報道,但生物分析軟件功能預(yù)測顯示可能具有致病性。推測可能為我國首例。

表2 患兒2基因檢測結(jié)果Table 2 Genetic test results of case 2 (9-month-old male child) of genic mutation
2 討論
精準(zhǔn)醫(yī)療是指在分子醫(yī)學(xué)技術(shù)的指導(dǎo)下,對某些診斷不明、療效不好的患兒,通過對DNA的檢測幫助醫(yī)生對疾病做出正確的診斷、治療及預(yù)后判斷,從而給患兒明確的治療方向,避免不必要的治療,給患兒家庭減輕負(fù)擔(dān)。
本文患兒1根據(jù)臨床癥狀及基因檢測確診為Sj?gren-Larsson綜合征,該病在世界上屬于罕見病例,發(fā)病率為0.4/100 000[1];是一種罕見的常染色體隱性遺傳性神經(jīng)魚鱗病樣綜合征。臨床以先天性魚鱗病、痙攣性雙下肢癱或四肢癱及智力障礙為特點[2]。對于本病的治療尚無其他治療方法,僅有支持療法對癥處理,運動落后者給予康復(fù)治療,長期的理療對于抵抗痙攣和保存活動能力非常重要[3];但是,康復(fù)治療效果不明顯,患兒不能獨立行走,只能借助輪椅生活;若無基因檢測,患兒常被誤診為痙攣型腦癱,長期在醫(yī)院康復(fù)治療,消耗患兒家屬大量時間,也造成嚴(yán)重的經(jīng)濟負(fù)擔(dān);本例患兒明確診斷后,家屬放棄在院治療,轉(zhuǎn)為家庭康復(fù)。建議臨床若懷疑Sj?gren-Larsson綜合征時,注意診斷兩個要點:皮膚干燥粗糙如魚鱗和痙攣性癱瘓,早期行基因檢測有助于及時發(fā)現(xiàn),明確國內(nèi)ALDH3A2的突變譜及為早期診斷、遺傳咨詢產(chǎn)前檢查提供依據(jù)。目前基因治療還存在許多問題,尚在試驗探索階段,然而基因治療是將來實現(xiàn)根治的期望。
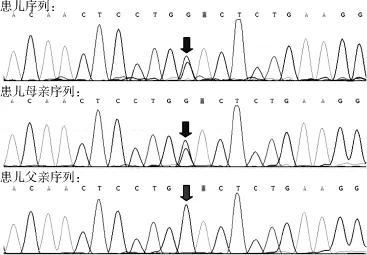
圖2 患兒2及其父母SUCLG1基因測序圖Figure 2 SUCLG1 genomic sequence of case 2 (9-month-old male child) of genic mutation and his parents shows the compound heterozygous for a mutation c.961C>G(p.A321P)coming from the mother
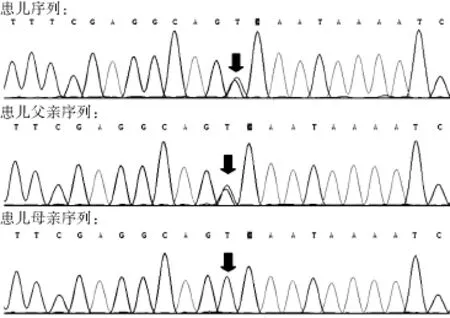
圖3 患兒2及其父母SUCLG1基因測序圖Figure 3 SUCLG1 genomic sequence of case 2 (9-month-old male child) of genic mutation and his parents shows the compound heterozygous for a mutation c.713T>C(p.D238 G)coming from the father
本文患兒2確診為線粒體DNA耗竭綜合征9型,其發(fā)病率至少為1/5 000[4],屬于線粒體疾病的一個類型;兒童線粒體腦病臨床表現(xiàn)多樣,容易誤診,陳健等[5]報道該院在2008—2013年共收治11例線粒體腦病患兒,其中7例曾被誤診,誤診率極高;其主要原因是不同患兒的同一點突變可出現(xiàn)不同的異常臨床癥狀[6];本例患兒早期曾誤診為腦損傷、腦性癱瘓,分別在多家醫(yī)院康復(fù)治療,療效不好,最后經(jīng)基因檢測確診為線粒體DNA耗竭綜合征9型。
線粒體DNA耗竭綜合征臨床上分為肌病、腦肌病、肝性腦病和神經(jīng)胃腸道疾病4種,目前發(fā)現(xiàn)由9種維持線粒體DNA穩(wěn)定性的核基因突變所致[7];其中腦肌病型(SUCLA2、SUCLG1、RRM2B)SUCLA2缺陷,全球約報道20例患兒。關(guān)于SUCLG1缺陷的研究較少,臨床表現(xiàn)與SUCLA2缺陷類似,但癥狀較重[8]。線粒體DNA耗竭綜合征9型由SUCLG1基因突變引起,以常染色體隱性方式遺傳,該型僅伴有甲基丙二酸尿癥,是由于SUCL缺陷,不能催化琥珀酰CoA生成琥珀酸,造成琥珀酰CoA堆積,同時使甲基丙二酰CoA生成琥珀酰CoA的過程受阻,導(dǎo)致甲基丙二酸升高[7];該患兒攜帶SUCLG1基因的兩個雜合突變:c.961C>G(p.A321P)和c.713T>C(p.D238G),其中c.961c>G(p.A321P)為文獻(xiàn)報道的已知致病性突變[8],而c.713T>C(p.D238G)未見文獻(xiàn)報道,因此,SUCLG1基因c.961C>G(母)和c.713T>C(父)2個雜合突變可能為我國首例,該報道擴展了SUCLG1基因突變譜,為患兒病因診斷及該家系的遺傳咨詢和產(chǎn)前診斷提供了分子依據(jù)。
總之,在既往的臨床工作中遺傳代謝性疾病多見,多以運動落后,發(fā)育遲緩,運動姿勢異常,語言障礙,癲癇,反復(fù)嘔吐或腹瀉,體質(zhì)量不增,肌張力障礙等癥狀而就診,若無基因檢測技術(shù)協(xié)助診斷,患兒難以明確診斷,僅是常規(guī)的康復(fù)手段治療,對一般發(fā)育落后,腦性癱瘓患兒來說,可以取得較好療效;而對于遺傳代謝性疾病來說,診斷難以明確,治療效果甚微,甚至延誤病情;目前隨著醫(yī)學(xué)的發(fā)展,分子遺傳學(xué)已被廣泛應(yīng)用于臨床,某些患有遺傳代謝性疾病的患兒能夠得到正確的診斷與治療,同時也給患兒明確的預(yù)后判斷;因此,基因檢測技術(shù)在臨床中顯得尤為重要,能夠為更多的患兒獲得準(zhǔn)確的治療和優(yōu)質(zhì)的服務(wù)。
本文無利益沖突。
[1]高蕾,蔣麗瓊,石秀玉,等.小兒Sj?gren-Larsson綜合征臨床特征及基因突變分析[J].中國實用兒科雜志,2014,29(6):458-461.DOI:10.7504/ek2014060614.GAO L,JIANG L Q,SHI X Y,et al.Analysis of clinical manifestations and gene mutations of Sj?gren-Larsson syndrome[J].Chinese Journal of Practical Pediatrics,2014,29(6):458-461.DOI:10.7504/ek2014060614.
[2]許汝釵,宋蘭萍,張英,等.Sjogren-Larsson綜合征一家系二例報告[J].中國優(yōu)生與遺傳雜志,2004,12(5):129.DOI:10.3969/j.issn.1006-9534.2004.05.075.XU R C,SONG L P,ZHANG Y,et al.Two cases report of Sjogren-Larsson syndrome[J].Chinese Journal of Birth Health and Heredity,2004,12(5):129.DOI:10.3969/j.issn.1006-9534.2004.05.075.
[3]張軼凡,黃堅.魚鱗樣紅皮癥-痙攣性截癱-智力發(fā)育不全綜合征(Sjogren-Larsson綜合征)(附1例報道及文獻(xiàn)分析)[J].齊齊哈爾醫(yī)學(xué)院學(xué)報,2013,34(22):3335-3336.ZHANG Y F,HUANG J.Sjogren-Larsson syndrome(one case report and literature review)[J].Journal of Qiqihar University of Medicine,2013,34(22):3335-3336.
[4]劉志梅,方方,丁昌紅,等.二代測序在兒童線粒體病診斷中的應(yīng)用[J].中華兒科雜志,2015,53(10):747-753.DOI:10.3760/cma.j.issn.0578-1310.2015.10.009.LIU Z M,F(xiàn)ANG F,DING C H,et al.Diagnosis of mitochondrial disorders in children with next generation sequencing[J].Chinese Journal of Pediatrics,2015,53(10):747-753.DOI:10.3760/cma.j.issn.0578-1310.2015.10.009.
[5]陳健,鄒麗萍.兒童線粒體腦病的臨床和分子遺傳學(xué)特點及其預(yù)后[J].臨床兒科雜志,2014,32(11):1020-1023.DOI:10.3969/j.issn.1000-3606.2014.11.006.CHEN J,ZOU L P.Clinical and molecular-genetic features and prognosis of mitochondrial encephalopathy in children[J].Journal of Clinical Pediatrics,2014,32(11):1020-1023.DOI:10.3969/j.issn.1000-3606.2014.11.006.
[6]黃圓圓.線粒體疾病研究進(jìn)展[J].國際兒科學(xué)雜志,2013,40(5):505-510.DOI:10.3760/cma.j.issn.1673-4408.2013.05.019.HUANG Y Y.Research progress of mitochondrial disease[J].International Journal of Pediatrics,2013,40(5):505-510.DOI:10.3760/cma.j.issn.1673-4408.2013.05.019.
[7]劉志梅,方方,丁昌紅,等.SUCLA2相關(guān)腦肌病型線粒體DNA耗竭綜合征一例并文獻(xiàn)復(fù)習(xí)[J].中華兒科雜志,2014,52(11):817-821.DOI:10.3760/cma.j.issn.0578-1310.2014.11.005.LIU Z M,F(xiàn)ANG F,DING C H,et al.SUCLA2-related encephalomyopathic mitochondrial DNA depletion syndrome:a case report and review of literature[J].Chinese Journal of Pediatrics,2014,52(11):817-821.DOI:10.3760/cma.j.issn.0578-1310.2014.11.005.
[8]劉玉鵬,李溪遠(yuǎn),丁圓,等.琥珀酰輔酶A連接酶缺陷導(dǎo)致繼發(fā)性甲基丙二酸尿癥四例的臨床與實驗室研究[J].中華 兒 科 雜 志,2016,54(5):365-368.DOI:10.3760/cma.j.issn.0578-1310.2016.05.011.LIU Y P,LI X Y,DING Y,et al.Clinical and laboratory studies on four Chinese patients with succinate-CoA ligase deficiency noticed by mild methylmalonic aciduria[J].Chinese Journal of Pediatrics,2016,54(5):365-368.DOI:10.3760/cma.j.issn.0578-1310.2016.05.011.
猜你喜歡
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
廣東醫(yī)科大學(xué)學(xué)報(2020年6期)2020-02-06 06:00:56
特別健康(2018年2期)2018-06-29 06:13:44
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
中國衛(wèi)生(2014年6期)2014-11-10 02:30:50
中國中醫(yī)藥現(xiàn)代遠(yuǎn)程教育(2014年23期)2014-03-01 04:33:45
