傣藥對葉豆葉中大黃酸的含量測定
2018-07-11 09:57:06卯明霞陸應彩姜明輝
中國民族醫藥雜志 2018年6期
卯明霞 陸應彩 彭 霞 姜明輝
(西雙版納州食品藥品檢驗所,云南 景洪 666100 )
傣藥對葉豆葉傣語“芽拉勐龍” ,《西雙版納傣藥志第二集》也稱 “牙郎滿聾”、翅葉槐;《云南思茅中草藥選》以翅決明、翅果決明、非州木通收載,為豆科植物翅莢決明Cassia alata Linn.的干燥葉。傣醫臨床常外用[1]于治療神經性皮炎,牛皮癬,溫疹,皮膚瘙癢,疔瘡腫瘍。文獻報道[2]含有:大黃酸、蘆薈大黃素、大黃素、蘆薈大黃素-8-O-β-D-葡萄糖苷、蘆薈大黃素-3-(羥甲基)-O-β-葡萄糖苷、-生育酚、羽扇豆-20(29)-烯-3-酮、β-谷甾醇、2-乙基-3-甲基-馬來酸酐、鄰二甲氧基對羥基-E-肉桂酸、2,3-二乙酰基丁四醇等化學成分,未見對葉豆葉大黃酸含量測定的有關報道,按《中國藥典》2015年版四部通則[3]現建立對葉豆葉大黃酸含量測定方法,為對葉豆葉質量評價傣醫臨床用藥提供參考方法和科學依據。
1 材料
1.1儀器Agilent高效液相色譜儀(G4214B-1260DAD;G1316A-1230TCC;G1329-1260ALS;G1311C-1260QuatpumpVL);島津UV-2550紫外可見光分光光度計;101-1EBS電熱鼓風干燥箱(北京市永光明醫療儀器廠);YB-1A真空恒溫干燥箱(天津市鑫洲科技有限公司);AE200電子分析天平(瑞士梅特勒);BL310電子天平(德國賽多利斯);MS205DU電子分析天平(梅特勒-托利多儀器(上海)有限公司)。
1.2試劑對照品大黃酸(中國食品藥品檢定研究院,批號:110757-201607)測定前在YB-1A真空恒溫干燥箱中以五氧化二磷干燥12h以上。乙腈、甲醇(賽默飛世爾科技(中國)有限公司)為色譜純,水為超純水(自制),其余試劑均為分析純。供試品對葉豆葉采自西雙版納州景洪市、勐海縣、勐臘縣。由西雙版納州食品藥品檢驗所彭霞主任藥師鑒定為豆科植物翅莢決明(Cassia alata Linn.)的干燥葉。
2 方法與結果
2.1色譜條件與系統適用性考察采用Waters Symmetry Shield RP18(150 mm ×4.6 mm,5.0 μm)色譜柱,流動相:乙腈-0.1%磷酸鹽緩沖液 (45 : 55)為流動相;流速:1 mL/min ;檢測波長:430 nm;柱溫:30℃;進樣量:10 μl;理論板數以大黃酸峰計不低于3000。色譜圖見圖1、圖2。
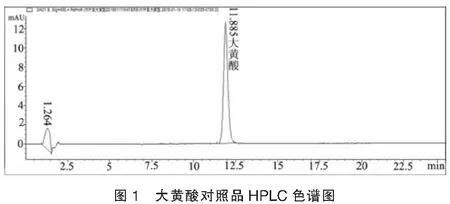
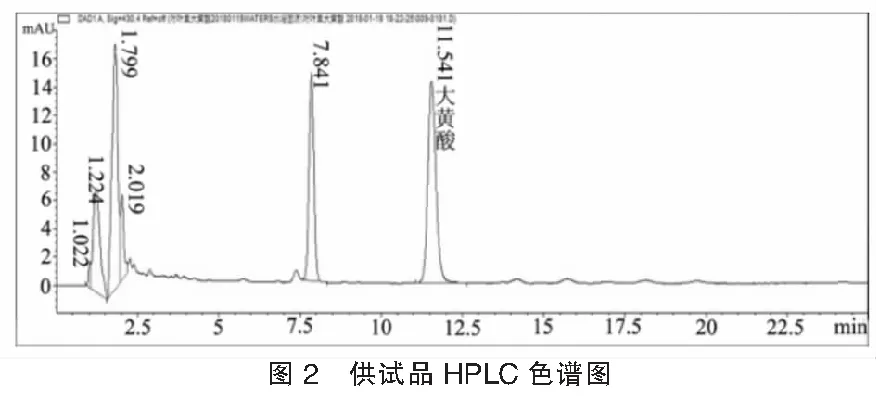
2.2溶液制備
2.2.1對照品儲備液的制備精密稱取大黃酸對照品適量(10.80mg),置于200mL量瓶中,用甲醇制成每1mL含54μg的溶液,搖勻,即得;(注:大黃酸在甲醇中溶解度較小,因此本實驗采用200mL的容量瓶,微加熱,即完全溶解)。
2.2.2供試品溶液的制備取供試品粉末0.5g,精密稱定,置錐形瓶中,精密加入甲醇溶液50mL,稱定重量,加熱回流1h(約70℃),放至室溫,再稱定重量,用甲醇溶液補足減失的重量,搖勻,濾過,取續濾液即得。
2.3方法學考察
2.3.1線性關系考察取濃度為54 μg / mL的對照品儲備液適量,加甲醇配制成1.35,2.70,5.40,16.20,21.60,32.40,48.60,54.00μg /mL的對照品溶液,搖勻。按上述色譜條件分別進樣10 μL ,以對照品濃度為橫坐標(X),對應色譜峰面積積分值為縱坐標(Y),繪制標準曲線,計算,得回歸方程:Y = 22.063x + 0.1869 ( r2=0.9998)結果表明:大黃酸對照品進樣量在1.35~ 54.00μg范圍內具良好的線性關系。
2.3.2重復性試驗取同一批次的對葉豆葉藥材(批號:20170820)6份,精密稱定,按“2.2.2”項下方法操作,依次測定,對葉豆葉中大黃酸的平均含量為0.65mg/g,RSD為0.70%。
2.3.3精密度試驗精密吸取大黃酸對照品溶液10μL,重復進樣7次,測得峰面積值的RSD為0.50%。
2.3.4穩定性試驗取同一批次(批號:20170820)的供試品溶液,分別在0,2,6,12,18,24 h進樣,測定峰面積,測得大黃酸峰面積的RSD為1.73%。表明供試品溶液在室溫下放置24h內穩定。
2.3.5加樣回收率試驗精密稱取已知含量的供試品(產地:西雙版納州景洪批號:20170820,含量:0.65mg/g)約0.25 g,平行6份,分別精密加入大黃酸對照品適量,按供試品溶液制備方法操作,測定,計算加樣回收率,見表1。
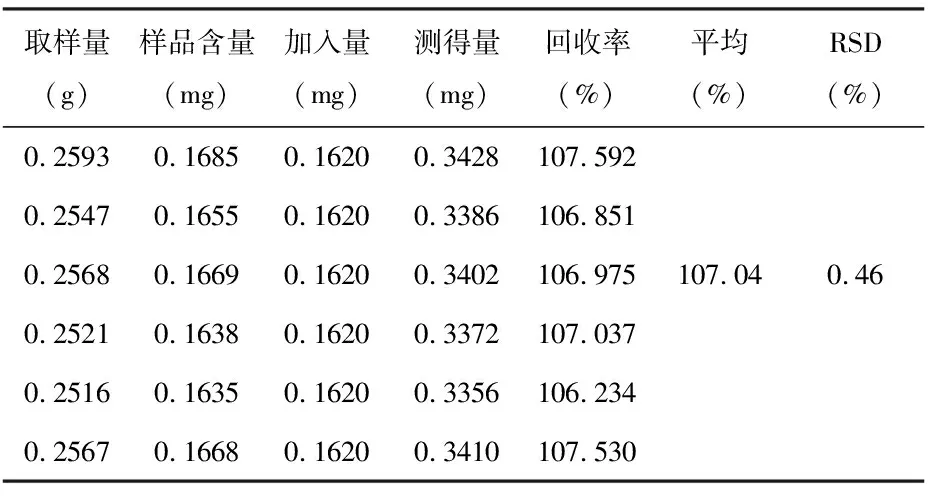
表1 加樣回收率試驗結果
3 樣品測定
按照上述色譜條件,分別測定5個不同產地5批供試品,計算含量,見表2。

表2 不同產地、不同批次的對葉豆中大黃酸的含量(n=2)
4 討論
本研究采用二極管陣列檢測器及紫外分光度計在190~450nm波長掃描,結果在254nm、430nm處有最大吸收,文獻報道[4]及《中國藥典》2015年版一部大黃項下,大黃酸檢測波長是430nm、 254nm,試驗中在254nm波長下的峰面積較大,但分離效果差,雜質峰影響大,在430nm波長下雜質成分干擾小,大黃酸峰的分離度大于1.5,確定檢測波長為430nm;流動相優選時分別以乙腈-0.1%磷酸(45:55)、乙腈-0. 1%冰乙酸(70 : 30)、甲醇-0.1%磷酸(85 : 15)、乙腈-0.1%甲酸(80:20)為流動相進行考察,結果顯示以乙腈-0.1%磷酸(45 : 55)為流動相,樣品分離度好,基線較穩定,最終確定此流動相。
本實驗考察了如下幾種供試品處理方法:乙醇、水、甲醇為溶劑,采用回流提取(提取時間為60min、90min、120min)、超聲處理(超聲時間為30min、45min、60min)。試驗結果表明,以甲醇為溶媒,回流提取60min對葉豆葉中大黃酸的含量較高,雜質干擾少。
另參照中國藥典2015年版四部“藥品質量標準分析方法驗證指導原則”(通則9101),考察了不同柱溫(25℃、30℃、35℃)、不同色譜柱:Dikma Diamonsil Plus C18 (250×4.6mm, 5.0 μm);Agilent ZORBOX SB- C18(150×4.6mm, 5.0 μm);Waters Symmetry Shield RP18(150 mm ×4.6 mm,5.0 μm);不同檢測波長(430nm、427nm、433nm);不同流速(0.8 mL/min、1mL/min 、1.2 mL/min)。結果顯示隨機變動因素對精密度的影響較小,進一步確保了試驗方法的科學性及數據的準確性,為對葉豆葉質量標準的進一步提高提供科學依據。
