兩親性殼聚糖基聚合物碳點的合成及其在載藥方面的應用
2018-07-11 03:21:42于淑娟朱永飛
發光學報 2018年7期
關鍵詞:殼聚糖
于淑娟,陳 寬,汪 豐,朱永飛
(廣西師范學院化學與材料科學學院,廣西南寧 530001)
1 引 言
熒光碳點是一種新型熒光碳納米材料,由于具有良好的水溶解性、生物相容性和低細胞毒性及穩定的光致發光性等優異性能,使其在生物成像、生物標記、光學催化、生物傳感器等領域有廣泛的應用前景[1-3]。碳點的原料來源廣泛,如:葡萄糖、檸檬酸、牛奶、柑橘等均可作為原料。隨著碳點的不斷深入研究,以非共軛線性聚合物如聚乙烯醇、聚丙烯酰胺、高分子多糖等為碳源制備的聚合物碳點及其應用成為另一研究熱點[4-7]。聚合物碳點與碳點相比具有易于提純、可保留聚合物的某些官能團、易于分子修飾,且與共軛熒光聚合物相比,具有水溶性好,制備簡單等優點。殼聚糖是一類天然高分子氨基多糖,具有無毒、成膜性好、易于分子修飾等諸多優點,已經廣泛應用于傳感器、生物醫用材料等方面。以殼聚糖為碳源合成聚合物碳點的研究也逐漸見諸報道,如文獻[8]通過水熱法合成了表面含有氨基的殼聚糖基聚合物碳點,熒光量子產率為7.8%。Xiao等[9]也利用殼聚糖為原料,采用微波法合成了殼聚糖基聚合物碳點。關于殼聚糖基聚合物碳點的應用范圍也在不斷擴大,如應用于熒光薄膜[10]、熒光涂層[11],光學催化[12]、離子檢測[13]等方面,但在藥物輸送研究中較少,另外,上述殼聚糖基聚合物碳點存在量子產率不高、活性位點相對較少、選擇性差等不足[14],這些缺陷也限制了它的應用性能。高量子產率是熒光材料的應用關鍵,設計合成高量子產率的聚合物碳點并進一步擴大其應用范圍具有重要意義。
熒光納米微球對于監測細胞識別腫瘤部位、監測藥物在組織內的運輸、確定治療等有重要作用[15],目前研究較多的一類是聚合物-無機雜化熒光納米微球,即聚合物包封半導體量子點,然而用于封裝的聚合物有導致量子點熒光消失的可能[16]。另一類是聚合物接枝熒光有機分子,但熒光有機分子通常有一定毒性。對于有實用價值的熒光納米微球,除了具有優良的熒光特性外,粒徑大小、分散性、表面修飾、生物毒性和生物相容性都是需要考慮的問題[17]。針對上述存在的不足,本研究首先合成了辛基化殼聚糖衍生物,然后接枝可以形成高量子產率碳點的檸檬酸小分子,并添加氮摻雜試劑N-(2-羥乙基)乙二胺,通過水熱法合成了具有高熒光量子產率(42.9%)的兩親性殼聚糖衍生物基聚合物碳點。并以阿霉素為模型藥物,將該聚合物碳點應用于藥物載體方面,對載藥及藥物釋放性能進行了研究,通過MTT法評價了載藥納米膠束的毒性以及對鼻咽癌的抑制效果。
2 實 驗
2.1 辛基殼聚糖(CS-g-OC)的制備
將2.0 g殼聚糖與一定量的辛醛加入到250 mL三口瓶中,加入50 mL甲醇,在氮氣保護下攪拌12 h,取1.5倍殼聚糖氨基物質量的硼氫化鉀溶于50 mL水中,滴加到上述混合溶液中,繼續反應12 h,結束后真空抽濾,用水和甲醇混合溶劑洗滌3次,冷凍干燥得到產物。辛基接枝率采用元素分析法進行計算。
2.2 辛基-檸檬酸殼聚糖(CS-g-OC-CA)的合成
取4.5 g檸檬酸溶于50 mL去離子水,加入1 g辛基殼聚糖攪拌12 h,然后加入0.02 mol的EDC。攪拌10 min 后,加入0.02mol的NHS,避光反應48 h,所得產物用去離水透析72 h(MWCO=8 000~14 000 u),然后冷凍干燥得到目標產物,合成路線如圖1(a)所示。
2.3 辛基-檸檬酸殼聚糖基兩親性聚合物碳點P(CS-g-OC-CA)Ds的制備
分別取上述不同辛基取代度的CS-g-OC-CA 0.25 g于水熱反應釜中,依次加入0.25 g無水檸檬酸、1 mL N-(2-羥乙基)乙二胺、8 mL去離子水進行充分混合,180℃反應3 h,結束后用去離子水透析72 h(MWCO=8 000~14 000 u),所得產物冷凍干燥。熒光量子產率測定根據參考文獻[18]的方法,以硫酸奎寧(0.05 mol/L H2SO4溶液中,360 nm激發光下,量子產率為0.54)為標準測定P(CS-g-OC-CA)Ds的量子產率,按式(1)進行計算:
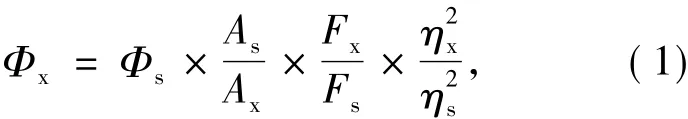
其中Φ是量子產率,F為測量的積分發射強度,A為光密度,η是溶劑的折射率。下標“s”是指具有已知量子產率的參考,“x”表示測定的P(CS-g-OC-CA)Ds。測得3種不同聚合物碳點Ⅰ、Ⅱ、Ⅱ的量子產率分別為 22.7%、42.9%、38.3%。
2.4 聚合物碳點載阿霉素(P(CS-g-OC-CA)Ds/DOX)納米膠束的制備
采用超聲法制備載藥納米膠束[19],取一定量P(CS-g-OC-CA)Ds溶于HCl與DMSO的混合溶液中(VHCl∶VDMSO=3∶7),超聲 30 min 后,40 ℃保溫12 h,加入一定量DOX的DMSO溶液,避光振蕩24 h后,用超純水透析72 h(MWCO=1 000 u)(每2 h換水一次),透析結束,用低溫10 000 r/min高速離心30 min除去上清液,得到P(CS-g-OC-CA)Ds/DOX納米膠束,其路線如圖1(b)所示。
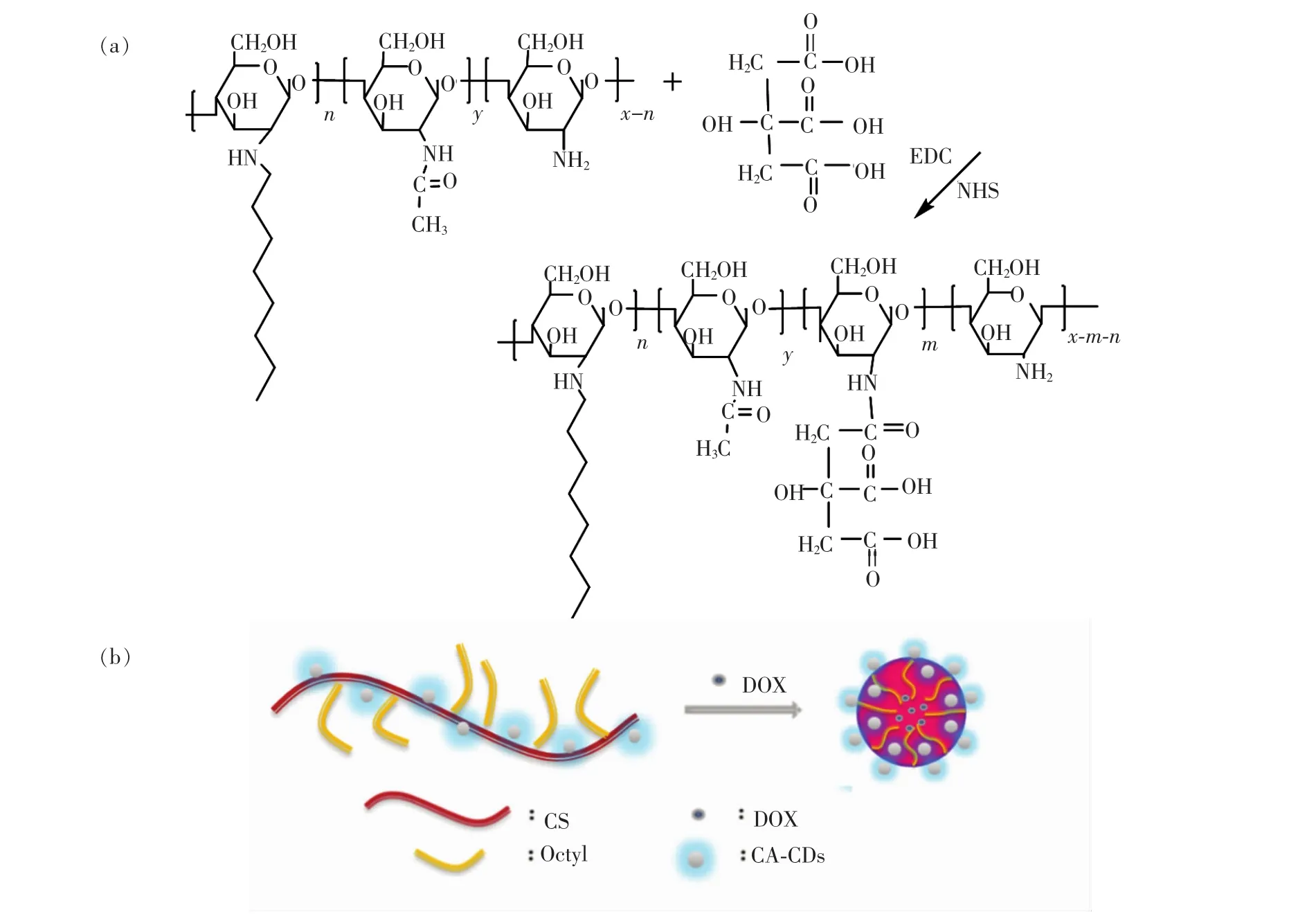
圖1 CS-g-OC-CA的合成路線(a)和P(CS-g-OC-CA)Ds/DOX的納米膠束示意圖(b)
采用IS10型FT-IR儀(美國NICOLET),以溴化鉀壓片法測定產物在500~4 000 cm-1范圍內的紅外光譜;采用美國Thermo Electron 250Xi型X射線光電子能譜分析P(CS-g-OC-CA)Ds表面化學組分,束斑尺寸為500μm。以日本島津公司RF-53-01對P(CS-g-OC-CA)Ds水溶液進行熒光光譜掃描,激發狹縫為3 nm,發射狹縫為3 nm。將P(CS-g-OC-CA)Ds/DOX納米膠束通過超聲分散后,采用英國馬爾文公司Zetasizer Nano ZS90動態光散射粒度儀測量其水合粒徑和電勢。采用美國TecnaiG2 F20 S-TWIN型透射電子顯微鏡觀察P(CS-g-OC-CA)Ds的形貌。采用日本JEOL公司JEM-2100高分辨率透射電子顯微鏡觀察P(CS-g-OC-CA)Ds/DOX的形貌。采用X射線衍射儀測定樣品的晶型結構,管壓為40 kV,掃描速率為2(°)/min,2θ為10°~45°。
3 結果與討論
殼聚糖衍生物及聚合物點的紅外譜圖如圖2所示。圖2(b)中CS-g-OC在2 926 cm-1和2 853 cm-1處特征峰與原料CS(圖2(a))對比明顯增強,為辛基中的亞甲基—CH2的特征吸收峰,說明辛基已經成功接到CS分子鏈上。圖2(c)中CS-g-OC-CA的紅外譜圖在3 465 cm-1與1 641 cm-1處吸收峰均明顯變寬,分別為檸檬酸中羧基—OH締合吸收峰與羰基C O的伸縮振動吸收峰,說明在辛基殼聚糖分子鏈上引入了檸檬酸。P(CS-g-OC-CA)Ds譜圖(圖2(d))中3 389 cm-1處寬峰為—OH、—NH2的伸縮振動特征峰,1 660 cm-1處為殼聚糖中酰胺—NH2的特征峰,1 590 cm-1為 N—H 的彎曲振動峰,1 076 cm-1處為—C—O—C的特征峰,從以上的分析中可知聚合物碳點表面含有—OH、環氧基、C O、—NH2等極性基團。
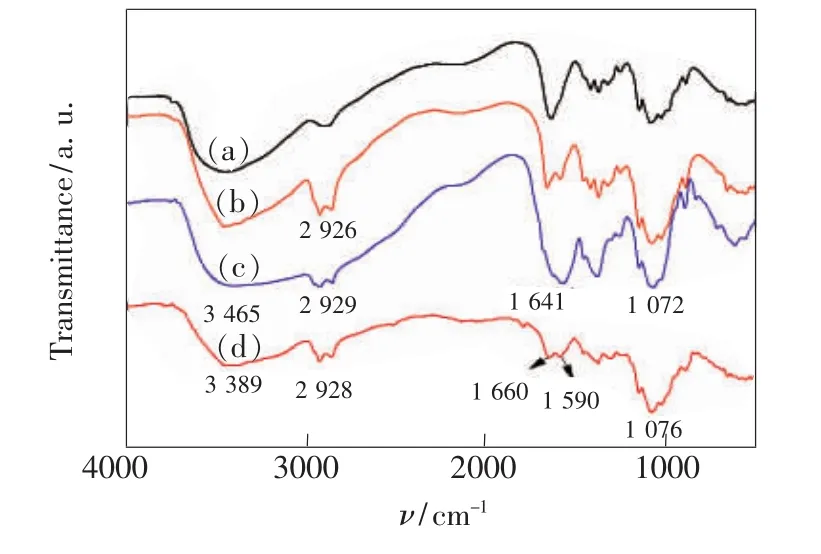
圖2 CS(a)、CS-g-OC(b)、CS-g-OC-CA(c)、P(CS-g-CA)Ds(d)的紅外光譜。
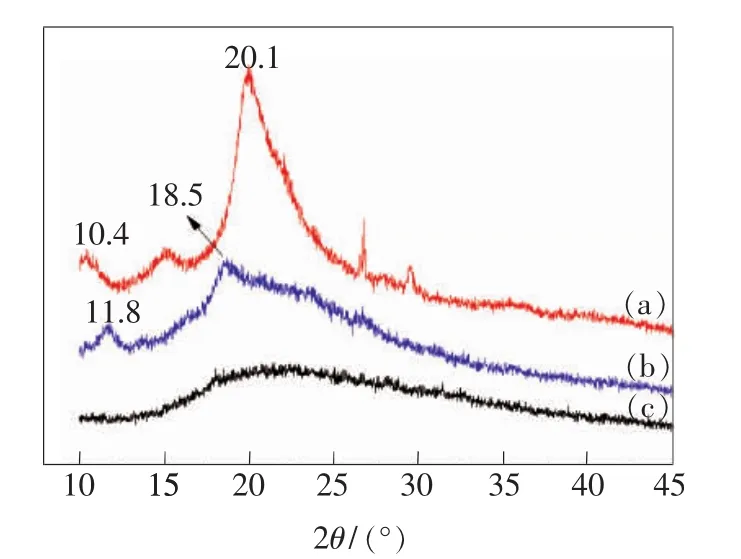
圖3 CS(a)、CS-g-OC-CA(b) 和 P(CS-g-OC-CA)Ds(c)的XRD圖。
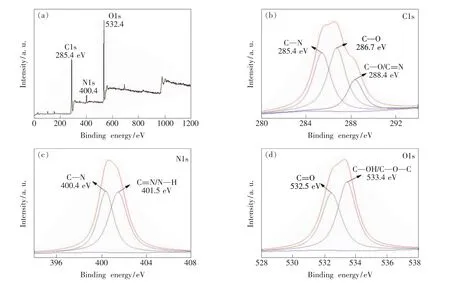
圖4 P(CS-g-OC-CA)Ds的 XPS圖譜及 C1s、N1s、O1s分峰。
為了進一步確定所合成的兩親性聚合物碳點結構并了解其晶型,采用X射線衍射(XRD)圖譜進行表征,如圖3所示。CS(圖3(a))在2θ=10.4°與20.1°處有強衍射峰,說明 CS具有高結晶性[20]。CS-g-OC-CA(圖3b)因辛基與檸檬酸的引入破壞了CS分子鏈的規整性,使結晶峰明顯減弱,在 11.8°和 18.5°處有弱結晶峰。在聚合物碳點P(CS-g-OC-CA)Ds(圖3(c))中,雖然檸檬酸發生了碳化分解,但辛基破壞CS分子鏈的規整性不變,使結晶峰成為漫散射衍射峰,說明聚合物碳點為無定型結構。
圖4為P(CS-g-OC-CA)Ds的X射線光電子能譜(XPS),圖4(a)中285.4,400.4,532.4 eV 分別為 C1s、N1s、O1s的 XPS譜,表明 P(CS-g-CA)Ds含有C、N、O 3種元素。在 C1s分峰譜圖中,285.4,286.7,288.4 eV 分別為 C—N、C—O、C O /C N的峰。N1s分峰中,400.4,401.5 eV分別為C—N、C N/N—H特征峰。O1s分峰中532.4 eV與533.4 eV分別為C O、C—OH/C—O—C的峰。以上分析表明該聚合物碳點表面含有—COOH、—OH、—NH2、環氧基等的功能基團,這與紅外譜圖信息一致,另外也正是因這些親水基團的存在使聚合物碳點表現出良好的水溶性。
P(CS-g-OC-CA)Ds的熒光光譜及紫外吸收光譜如圖5所示。P(CS-g-OC-CA)Ds在232 nm與348 nm處有紫外吸收,分別歸屬于聚合物碳點中 sp2碳的 π-π*躍遷和羰基的 n-π*躍遷[21],P(CS-g-OC-CA)Ds的最佳熒光激發峰與發射峰分別為376 nm和455 nm(圖5(a))。當激發波長從280~400 nm以間隔20 nm增加時,其發射光譜熒光表現出先增強后減弱的變化趨勢(圖5(b))。激發波長為360 nm時,熒光強度達到最大值,而且在整個過程中有紅移的現象,說明該聚合物碳點具有多色熒光性。熒光激發依賴發射行為可能歸因于聚合物碳點不同的表面態和尺寸[22],不同的表面態提供了多種電子遷移途徑和能級差[23]。另外尺寸的不同決定了其能量帶隙的不同,進而決定了聚合物碳點的發射位點也不同,多種不同發光中心使聚合物碳點具有熒光激發依賴性[24]。以硫酸奎寧為參比,測試了 P(CS-g-OCCA)Ds的熒光量子產率最高為42.9%,與純殼聚糖聚合物碳點[8-9]的量子產率相比明顯提高,可能歸因于殼聚糖分子與氮摻雜試劑在熱解過程中對檸檬酸碳點起到了鈍化作用,增加了聚合物碳點表面的缺陷,另外殼聚糖也是碳源材料,在熱解過程中也有部分殼聚糖基聚合物點的生成,以上綜合結果導致了熒光量子產率的增加。
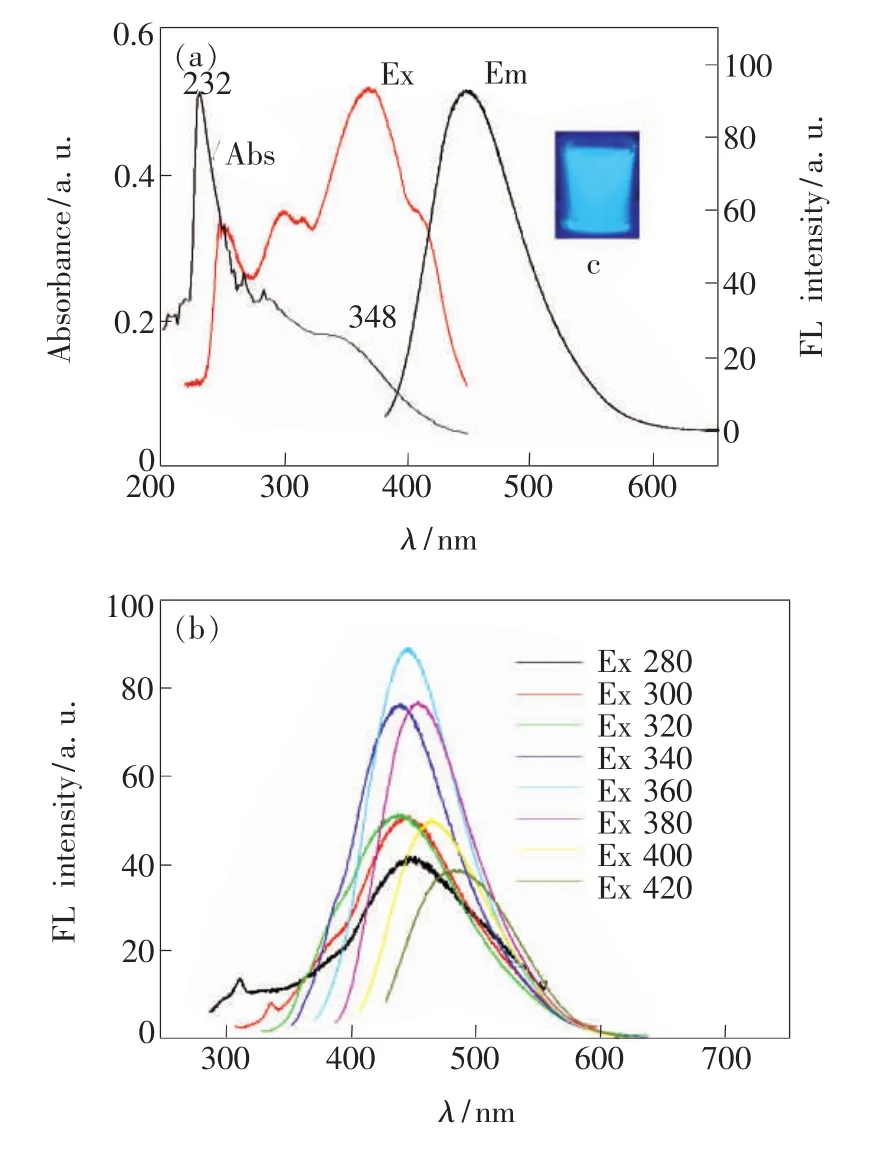
圖5 (a)P(CS-g-OC-CA)Ds的紫外吸收、熒光光譜,插圖:356 nm紫外光下的數碼照片;(b)不同激發波長下的發射光譜。
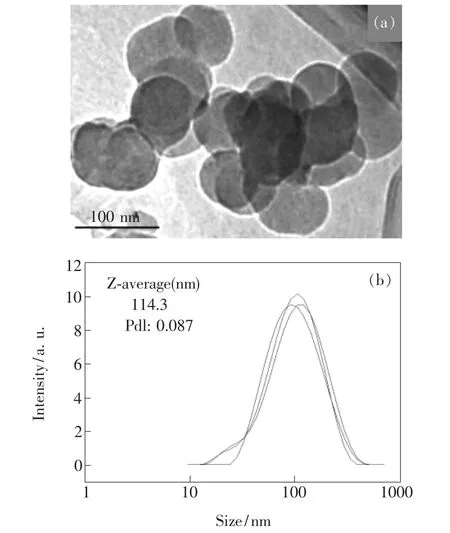
圖6 載藥納米膠束P(CS-g-OC-CA)Ds/DOX的高分辨透射電鏡圖(a)和流體力學粒徑分布曲線(b)
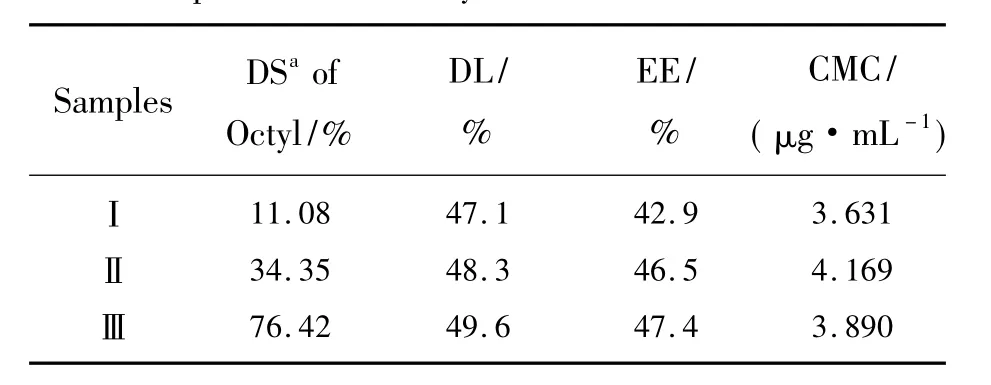
表1 不同取代度納米膠束的臨界膠束濃度、載藥量和包封率Tab.1 Criticalmicelle concentration,drug loading and encapsulation efficiency for differentmicelles
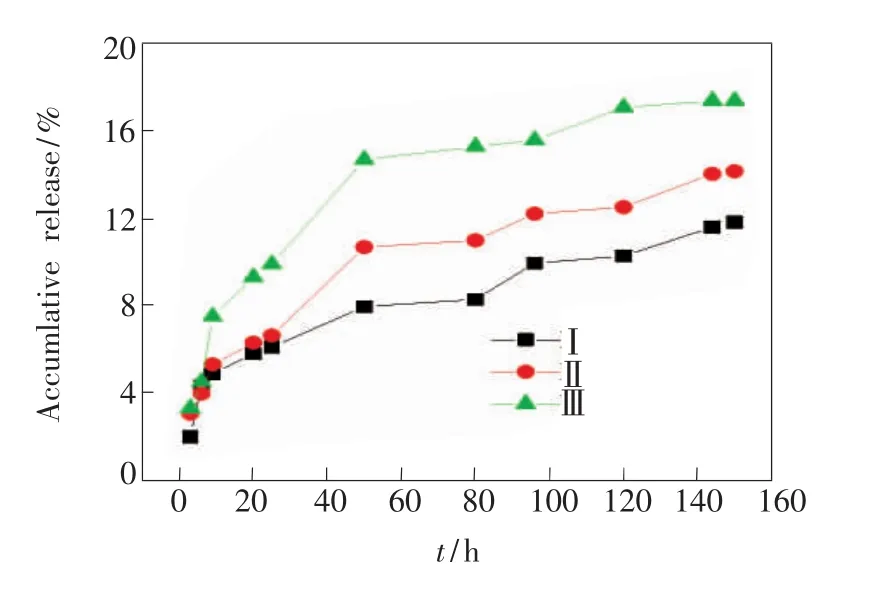
圖7 P(CS-g-OC-CA)Ds/DOX納米膠束的體外釋藥曲線(pH=7.4)
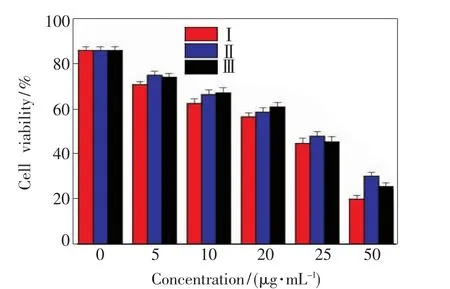
圖8 添加不同濃度P(CS-g-OC-CA)Ds/DOX對CNE-2細胞活性的影響
圖6為P(CS-g-OC-CA)Ds/DOX高分辨透射電鏡(圖6(a))與流體力學粒徑及其分布(圖6(b)),從HRTEM圖中可見P(CS-g-OC-CA)Ds/DOX在溶液中呈近似球型分布,粒徑約為80~90 nm,流體力學粒徑為114.3 nm,比透射電鏡測得的粒徑要大,可能是因為聚合物碳點表面含有羥基,羧基、氨基等官能團,導致納米膠束吸附在一起形成較大的團聚體使DLS的值較大。PDI為0.087,說明粒徑分布較窄,表面電勢為+5.72mV。對聚合物碳點載藥與釋藥性能進行了研究,其中聚合物碳點的臨界膠束濃度、以及對DOX的載藥量與包封率數據如表1所示。聚合物碳點當辛基取代度為76.42%時,有最佳的對DOX的載藥量和包封率,分別為49.6%與47.4%。藥物的體外釋放性能如圖7所示。可以看出,不同辛基取代度的聚合物碳點納米膠束中DOX的釋放行為均具有初期爆釋、后期緩慢釋放的雙相特點,在50 h釋放率分別為7.92%、10.64%、14.63%,隨后藥物釋放率有所減緩,不同辛烷基取代度對釋藥性能影響明顯,隨著辛烷基取代度的增加,釋藥性能增加,150 h后,3種取代度的聚合物碳點膠束的釋藥率分別達到11.77%、14.09%、17.31%。DOX 的緩慢釋放與包封機理相關,可能有部分DOX是吸附在聚合物碳點納米膠束表面,最初的10 h的爆釋歸因于吸附在納米膠束表面的DOX發生的解吸附。隨后DOX隨納米粒子的溶脹逐漸從內部擴散出來。后續的緩慢釋放可能是因為DOX被殼聚糖聚合物碳點中辛烷基的纏繞、包裹等結合得比較緊密導致,另外釋藥性也與載藥量有關,隨著載藥量的增加,DOX的釋放量也隨之增加。
將不同 DOX 含量(0,5,10,20,25,50 μg/mL)的載藥納米膠束P(CS-g-OC-CA)Ds/DOX與人鼻咽癌細胞CNE-2共同培養72 h,觀察其對細胞活性的影響,結果如圖8所示。未載藥納米膠束處理72 h后,細胞存活率仍在85%以上,表明辛基殼聚糖聚合物碳點基本沒有毒性。而負載DOX的納米膠束對鼻咽癌細胞表現出了明顯增強抑制效果,說明該載藥納米膠束在癌癥治療方面具有潛在的應用前景。
4 結 論
通過水熱法制備了一種辛基化殼聚糖基兩親性聚合物碳點熒光材料,發現檸檬酸的接枝可以明顯提高聚合物點的量子產率。該聚合物碳點以無毒生物相容性良好的殼聚糖為主要原料,使碳點保留了部分殼聚糖的性能。在阿霉素載藥方面的應用結果表明,該聚合物碳點在辛基取代度為76.42%時,其最大載藥量和包封率分別為49.6%與47.4%。另外我們還發現,通過控制辛基取代度可以對藥物的載藥量和包封率進行調控。MTT性能研究發現,未載藥納米膠束處理72 h后,細胞存活率仍在85%以上,表明辛基殼聚糖聚合物碳點基本沒有毒性,載藥納米膠束對鼻咽癌細胞有一定的抑制作用。由此可見,該聚合物碳點熒光材料不僅可以載藥還具有熒光示蹤的性能,是一類良好可示蹤的藥物載體材料。該聚合物碳點材料在生物醫學成像方面也將有很好的應用前景,可以為疾病診斷與治療提供支持,同時還可以利用聚合物碳點易于修飾的優點在表面連接靶向分子制備多功能醫用納米粒子,進一步擴大其應用范圍。
猜你喜歡
河北科技師范學院學報(2022年2期)2022-08-26 08:55:40
河北科技師范學院學報(2021年1期)2021-05-10 03:34:20
中成藥(2017年12期)2018-01-19 02:06:57
電源技術(2017年1期)2017-03-20 13:37:59
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
天然產物研究與開發(2016年1期)2016-06-05 10:29:25
食品界(2016年4期)2016-02-27 07:36:46
中國果菜(2015年2期)2015-03-11 20:01:01
應用化工(2014年7期)2014-08-09 09:20:21
應用技術學報(2014年4期)2014-02-28 14:52:40
