MDI合成聚氨酯型不飽和樹脂及其性能
2018-08-20 01:41:12錢建坐鎮
材料科學與工程學報 2018年4期
, ,,,錢建,坐鎮,
(1.華東理工大學材料科學與工程學院,特種功能高分子材料及相關技術教育部重點實驗室,上海 200237;2.華東理工大學華昌聚合物有限公司,上海防腐蝕新材料工程技術研究中心,上海 200237)
1 前 言
不飽和樹脂通常是由二元醇、飽和二元酸及不飽和二元酸合成的一種熱固性樹脂,具有較低的成本、優異的浸潤性、良好的可加工性,但不飽和樹脂較低的沖擊強度和斷裂延伸率、較大的固化收縮率等缺點限制了其應用[1-4]。為了提高不飽和樹脂的性能,Benny Cherian向不飽和樹脂中加入環氧樹脂來改進不飽和樹脂的力學性能[5]。李揚俊借鑒聚氨酯丙烯酸酯(PUA)的合成方法以二元醇、二元酸酐和甲苯二異氰酸酯(TDI)為原料合成了一種聚氨酯型不飽和聚酯樹脂(UPPU),樹脂鏈段上同時含有不飽和鏈節和聚氨酯基團,一定程度上綜合了兩者的性能達到取長補短的目的[6-14]。但是,關于聚氨酯型不飽和聚酯樹脂的研究報道還比較少。
本文以二苯基甲烷二異氰酸酯、甲基丙烯酸羥乙酯、一縮二乙二醇、鄰苯二甲酸酐和順丁烯二酸酐為原料合成了聚氨酯型不飽和聚酯樹脂,然后加入交聯劑苯乙烯或甲基丙烯酸甲酯,制備了室溫下為液態的熱固性樹脂,再加入過氧化物類引發劑制得室溫下成固態的材料,并研究了樹脂的力學性能、熱穩定性。
2 實驗部分
2.1 主要原料與儀器
二苯基甲烷二異氰酸酯(MDI)、甲基丙烯酸羥乙酯(HEMA)、馬來酸酐、鄰苯二甲酸酐、一縮二乙二醇、一縮二丙二醇、苯乙烯、環烷酸鈷都為工業級。叔丁基鄰苯二酚、甲基苯醌為化學純。MERICAN 9708:自制。
2.2 聚酯二元醇的制備
首先,在裝有攪拌、熱電偶、冷凝管的三口燒瓶中加入一縮二乙二醇和二元酸,初始溫度為160℃,邊攪拌邊反應,反應一段時間后升溫,升溫過程要精確控制餾頭溫度,使其低于105℃,避免反應過程中醇的揮發;測定不同反應時刻的酸值,確定反應程度,當酸值達到10mg/g時可認為反應完全;通過此反應制備了R1、R2兩種聚酯二元醇。反應方程式如圖1(a)所示,R1通過鄰苯二甲酸酐和一縮二乙二醇之間的反應制備,R2通過馬來酸酐和一縮二乙二醇之間的反應制備,調節R1、R2的比例可得到不同分子結構的樹脂體系。
2.3 聚氨酯預聚物的制備
MDI和HEMA加入帶攪拌的三口燒瓶,插入熱電偶,反應溫度為70℃±5℃,反應過程中通過化學滴定法測定不同時刻異氰酸根(-NCO)的含量從而確定反應程度,將體系中-NCO含量降至反應初始值一半的時刻定為反應終點,此刻終止反應,即制得聚氨酯預聚物。反應方程式如圖1(b)所示。
2.4 聚氨酯型不飽和聚酯樹脂(UPPU)的制備
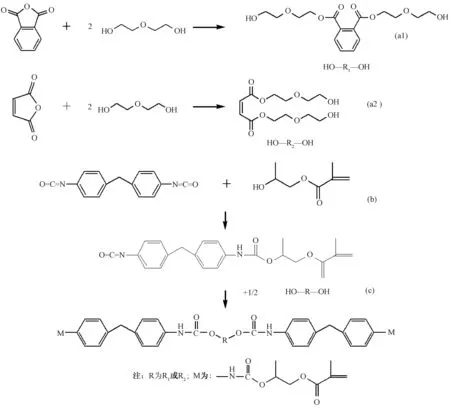
圖1 聚氨酯型不飽和樹脂的制備反應式 Fig.1 Schematic for the synthesis of unsaturated polyester polyurethane resin (UPPU)
在三口燒瓶中進行聚氨酯預聚物和聚酯二元醇的反應,反應方程式如圖1(c)所示,反應溫度為70℃±5℃。為防止反應過程中粘度過大,反應前需加入低粘度交聯劑。反應過程中利用傅里葉-紅外光譜分析儀檢測不同時刻-NCO特征吸收峰的強度從而判斷反應的進行程度,由于該吸收峰強度與反應程度呈負相關,所以當-NCO特征吸收峰消失時,表明反應結束。體系達到反應終點后加入適量交聯劑和阻聚劑,使樹脂黏度降至500cp左右,收集待用,由此制備出聚氨酯型不飽和聚酯樹脂。
通過上述步驟制備了五種樹脂體系,如表1所示。以苯乙烯為交聯劑,改變二元醇投料比(R1∶R2)制備了1#、2#、3#三組樣品;在聚酯二元醇投料比(R1∶R2)為1∶1的情況下,改變交聯劑種類制備了4#和5#兩組樣品。

表1 聚氨酯型不飽和聚酯樹脂體系的組成Table 1 Composition of UPPU
2.5 測試表征
通過測定聚酯二元醇合成過程中不同時刻的酸值測定聚酯二元醇的反應程度,酸值測試參照GB/T 7193-2008。測定聚氨酯預聚物合成過程中的的異氰酸根含量,得到預聚物的反應程度,測試方法參照HG/T 2409-92。采用傅里葉-紅外光譜分析(FT-IR),測試不同反應時刻的-NCO吸收峰強度,從而定性判斷縮聚反應程度。利用Nicolet IS5型FT-IR透過模式,涂膜法進行紅外光譜分析。采用NDJ-7型旋轉粘度計測定穩態UPPU剪切黏度,測試標準參照GB/T 7193-2008。參照GB/T 2567-1995,在液態熱固性樹脂中加入過氧化甲乙酮(1.5wt%)和環烷酸鈷(1.5wt%),充分攪拌、除去氣泡后慢慢澆注到玻璃模中制備樹脂澆注體;然后,參照GB/T 2567-2008利用樹脂雕刻機將樹脂板材切割成標準樣條(拉伸強度的樣條規格為:長200mm啞鈴窄段寬10mm厚度4mm;彎曲強度樣條規格為:長80mm寬15mm厚度4mm;沖擊強度的樣條規格為:長80mm寬15mm厚度4mm);利用INSTRON 3382型萬能試驗拉伸機進行力學性能測試。利用Q20型DSC測定固化后樹脂的玻璃化轉變溫度(Tg),升溫速率為5℃/min,氮氣流。通過TGA(Q50,TA)測試固化后的樹脂的熱分解溫度(Td),升溫速率為10℃/min,氮氣流。
3 結果與討論
3.1 異氰酸根(-NCO)含量分析
圖2是聚氨酯預聚物制備過程中-NCO含量隨反應時間的變化曲線。從圖中可知,反應初始時刻-NCO含量在14%左右;反應開始后,曲線斜率呈兩段式,反應初期(反應3h)-NCO的含量隨反應進行迅速下降,3h后-NCO含量顯著低于初始值且隨反應時間的延長幾乎不再變化。異氰酸根和羥基(-OH)先快后慢的反應速率是因為-NCO和-OH的反應活化能低,導致初期反應速率快,且反應過程中放出的大量熱會進一步加速反應的進行,但隨反應時間的延長有效基團含量降低,反應速率隨之減慢,所以為防止前期反應過快,需嚴格控制反應溫度。另外,反應4h后,-NCO含量降為初始值的一半,此時即可說明異氰酸根反應掉一半,由圖1b反應方程式可知異氰酸酯根反應一半,即可認為聚氨酯預聚物制備完成。

圖2 聚氨酯預聚物制備過程中異氰酸根(-NCO)含量隨反應時間的變化曲線Fig.2 Content of isocyanato (-NCO) vs reaction time curve during the synthesis of polyurethane prepolymer
3.2 紅外光譜分析

圖3 聚氨酯型不飽和樹脂制備過程中-NCO紅外吸收峰強度隨反應時間的變化Fig.3 FTIR spectrum of content of isocyanato (-NCO) vs reaction time during the synthesis of UPPU
圖3是聚氨酯型不飽和樹脂制備過程中不同反應時刻的紅外譜圖。2270cm-1處是-NCO特征峰[15]。由圖3可知,在70℃、未加催化劑的反應條件下,隨縮聚反應時間的延長,-NCO紅外吸收峰強度逐漸降低,且反應5h后-NCO紅外吸收峰已基本消失,可認為異氰酸根全部參加了反應且反應完全,由此可知5h后反應基本完成;再加入其余的稀釋劑和適量阻聚劑,由此就制得聚氨酯型不飽和樹脂。
3.3 樹脂固化工藝分析
表2所示為聚氨酯型不飽和樹脂的固化工藝和黏度。以己二醇二丙烯酸酯(5#)為交聯劑時樹脂的黏度最大,這是因為己二醇二丙烯酸酯單體的黏度比苯乙烯和甲基丙烯酸甲酯的黏度大。不飽和樹脂使用的黏度范圍為200~600MPa·s,所以,除5#樹脂外,其它樹脂體系的黏度適中,比較適合加工。從樹脂的凝膠時間可見,合成樹脂的凝膠時間要長于商品化的9708,可能是因為合成樹脂后期加入的較多阻聚劑減緩了凝膠化反應,因此可以通過改變阻聚劑的加入量來調節凝膠時間。樹脂在25℃的恒溫固化過程中,3#樹脂的放熱峰溫度高于其它聚氨酯型不飽和樹脂,原因是其R2(馬來酸酐)的含量高,導致樹脂體系中雙鍵含量高于其他幾個型號的樹脂,而固化反應為雙鍵的加聚反應,加聚反應放出的熱量在固化過程中積累,導致固化過程中溫度高于其它樹脂。
3.4 力學性能分析
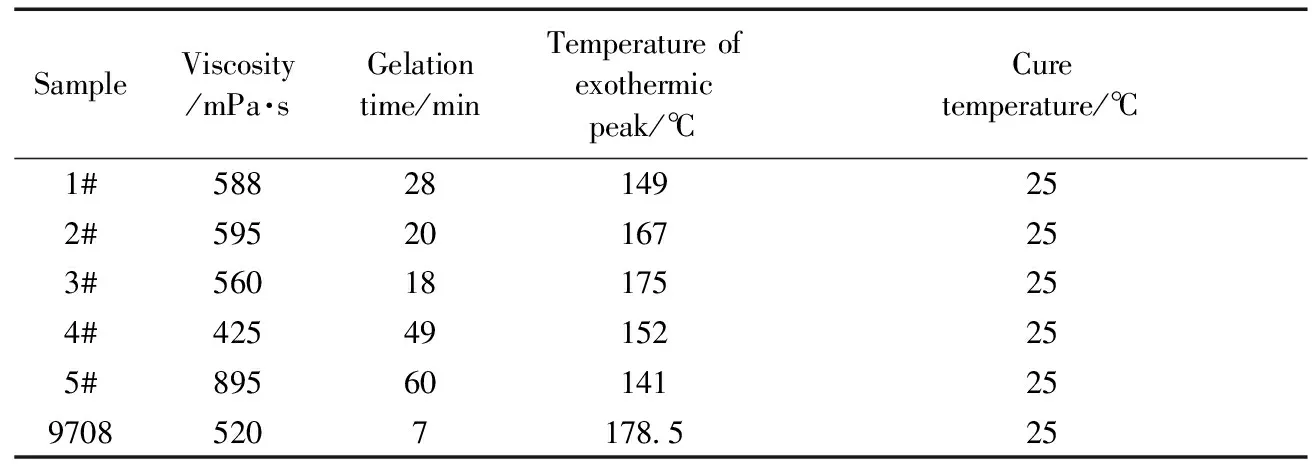
表2 樹脂的固化工藝Table 2 Resin curing process
由圖4可知,聚氨酯型不飽和樹脂的力學性能,如2#樹脂的拉伸強度、彎曲強度、沖擊強度和斷裂延伸率,優于通用型不飽和樹脂(9708),且分別提升26%,42%和270%。這是因為聚氨酯型不飽和樹脂中存在分子間的氨基甲酸酯氫鍵作用,而通用型不飽和樹脂中存在分子間酯鍵的相互作用,前者內聚能為36.4kJ/m2,是后者內聚能(12.1kJ/m2)的三倍,高內聚能的氨基甲酸酯鍵增強了分子間作用力從而提高了不飽和樹脂的力學性能。當R1∶R2為1∶1時,交聯劑為甲基丙烯酸甲酯(4#)或苯乙烯(2#)的樹脂體系其拉伸強度、彎曲強度及沖擊強度均明顯高于己二醇二丙烯酸酯為交聯劑的樹脂體系(5#),可能是因為5#樹脂分子結構中的兩個不飽和雙鍵使得樹脂固化后增大了材料的交聯點密度,使其脆性增大,從而使斷裂延伸率和沖擊強度下降。4#和2#相比,兩者的彎曲強度、沖擊強度相近,但2#的拉伸強度和斷裂延伸率低于4#;4#的彎曲模量高于2#和5#,表明由甲基丙烯酸甲酯制備的材料的剛性要優于其余二者。交聯劑為苯乙烯時,當分子結構全部為R1(鄰苯二甲酸酐),即1#樹脂,由于R1為鄰苯二甲酸酐能提高樹脂分子主鏈上苯環的比例,而苯環的剛性會抑制材料在外力作用下的形變、降低材料延展性;并且苯環的剛性又會降低材料的抗沖擊性能,導致1#樹脂沖擊強度和斷裂延伸率低于2#和3#。當R1∶R2為1∶1,即2#樹脂時,材料的拉伸強度、彎曲強度和沖擊強度最大,分別為76MPa,155MPa和25.00kJ/m2,原因在于該樹脂體系中R1、R2的含量各占一半且綜合了兩者性能,從而使2#樹脂具有最高的力學強度和最好的韌性。
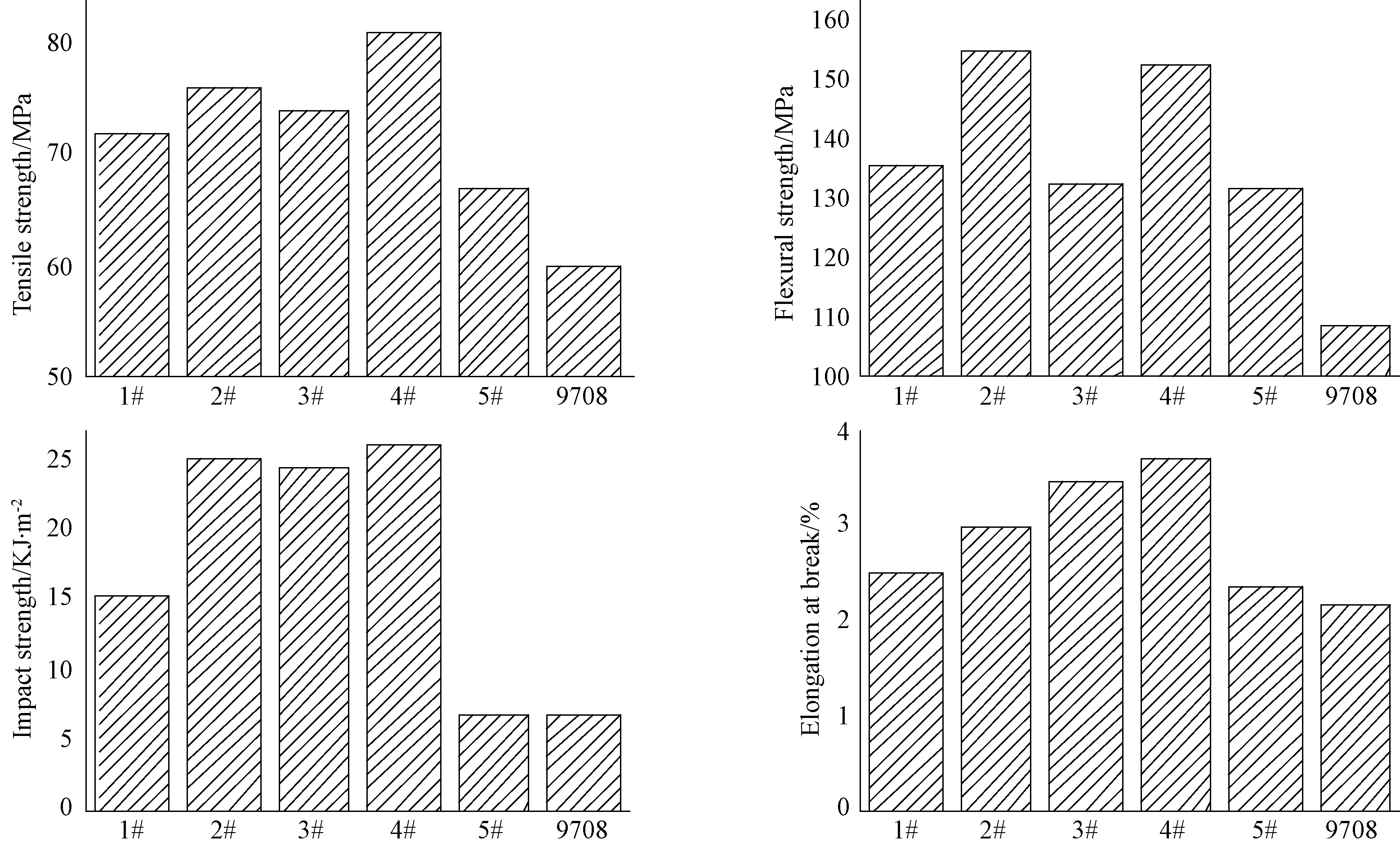
圖4 聚氨酯型不飽和樹脂與通用型不飽和樹脂的力學性能的比較Fig.4 Mechanical properties ofUPPU and universal unsaturated resin
3.5 DSC分析
圖5是采用不同交聯劑制備的樹脂的DSC曲線。從圖5可知,苯乙烯、甲基丙烯酸甲酯和己二醇二丙烯酸酯為交聯劑時,聚氨酯型不飽和樹脂的玻璃化轉變溫度(Tg)分別為115℃,120℃ 和107℃,由此可知甲基丙烯酸甲酯作為樹脂稀釋劑和交聯劑時樹脂的Tg最高;苯乙烯和己二醇二丙烯酸酯作為稀釋劑和交聯劑時樹脂的Tg均高于100℃,表明樹脂具有良好的耐熱性。己二醇二丙烯酸酯分子骨架上有較長的烷基鏈,使規整性降低、自由體積增大,導致烷基鏈鍛更易發生自由運動從而降低了材料的Tg。聚甲基丙烯酸甲酯均聚物的Tg(105℃)高于聚苯乙烯均聚物的Tg(100℃),因此單體甲基丙烯酸甲酯作為樹脂交聯劑使用時,樹脂固化物的Tg會高于苯乙烯單體,但是二者的差別不是很大。
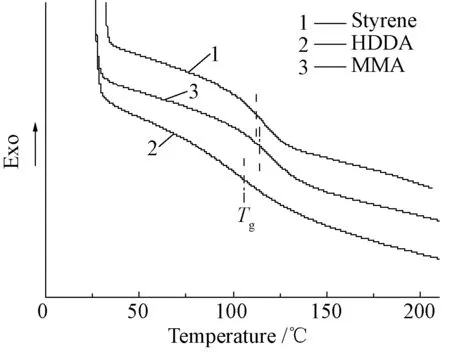
圖5 不同交聯劑材料的DSC曲線Fig.5 DSC curves of UPPU with different corsslinkers
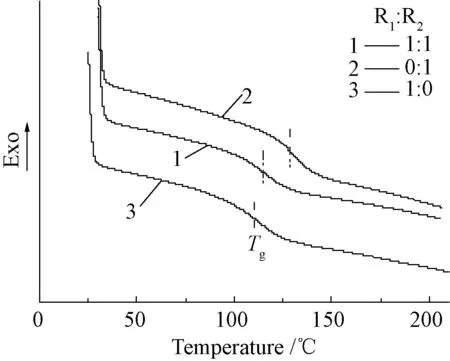
圖6 不同R1、R2比例下聚氨酯型不飽和樹脂的DSC曲線Fig.6 DSC curves of UPPU with different molar ratio of R1 and R2
圖6是以苯乙烯為交聯劑,改變R1和R2比例制備的樹脂的DSC曲線。從圖6中可知,當R1∶R2為0∶1時,不飽和樹脂的Tg最大為130℃;當R1∶R2為1∶0時,不飽和樹脂的Tg為110℃;當R1∶R2為1∶1時,不飽和樹脂的Tg介于二者之間為115℃。由此可知不飽和樹脂的Tg會隨著分子骨架中馬來酸酐比例的增加而升高;原因在于馬來酸酐中的雙鍵能夠增大固化樹脂的交聯點密度從而使鏈鍛運動進一步受阻,導致Tg升高,從而拓寬了不飽和樹脂的使用溫度范圍。
3.6 TGA分析
圖7是苯乙烯為交聯劑、不同R1、 R2比例下制備的樹脂的熱分解曲線。從圖7可知,聚氨酯型不飽和樹脂(1#、2#和3#)的熱分解溫度(415℃)均高于通用不飽和樹脂(9708,400℃),表明聚氨酯型不飽和樹脂具有更好的熱穩定性;而不同R1、R2比例下的樹脂的熱分解溫度大致相同,則說明鄰苯二甲酸酐或者馬來酸酐含量的差別對樹脂的熱分解溫度影響較小。
本文以二苯基甲烷二異氰酸酯(MDI)、丙烯酸羥乙酯、自制二元醇為原料合成了力學性能和熱性能均優于通用型不飽和樹脂的聚氨酯型不飽和樹脂,并研究了鄰苯二甲酸酐和馬來酸酐的配比(R1∶R2)、交聯劑種類對不飽和樹脂力學性能以及熱穩定性的影響。研究發現聚氨酯型不飽和樹脂的熱分解溫度高于通用不飽和樹脂,馬來酸酐含量的增加既可提高不飽和樹脂的斷裂延伸率又可提高樹脂的玻璃化轉變溫度,從而拓展了不飽和樹脂的使用溫度上限;另外,R1、R2的摩爾比為1∶1,甲基丙烯酸甲酯為交聯劑時,得到的UPPU力學性能最好,拉伸強度、彎曲強度、沖擊強度分別為81MPa、153MPa、26kJ/m2,是通用型不飽和樹脂(9708)的1.4、1.4、3.8倍。
