基因dat對單核細胞增生李斯特氏菌的生物學特性影響
2018-08-23 07:23:36曾海娟謝曼曼丁承超劉武康董慶利
微生物學雜志 2018年3期
關鍵詞:李斯特
曾海娟, 謝曼曼, 丁承超, 劉武康, 董慶利, 劉 箐
(上海理工大學 醫療器械與食品學院,上海 200093)
單增李斯特菌(Listeriamonocytogenes,Lm)是革蘭染色陽性的一種重要的食源性致病菌,廣泛分布于土壤、污水等環境中[1]。Lm具有較強的生存能力,pH在4.5~9.0,溫度在0~45 ℃均可增殖[2],在冰淇淋、原奶、生肉、干酪、海鮮及方便食品,如熟肉、熏魚中均有檢出[3-6]。由于食用了被污染的食物導致單增李斯特菌的感染[7],患者死亡率可達30%以上。單增李斯特菌是典型的細胞內寄生菌,可通過李斯特菌溶血素等的裂解作用下逃離吞噬小體[8-9],進入并在細胞質中增殖,在其中產生的蛋白將激活CD8+T細胞,部分被吞噬溶酶體直接殺死,降解的蛋白呈遞給CD4+T細胞。Lm引起的獨有的這種功能免疫應答機制,使得減毒李斯特菌成為一種潛在的可攜帶腫瘤、病毒及細菌抗原的疫苗載體[10]。 基因dat(daaA)存在于多種細菌中,其編碼D-丙氨酸氨基轉移酶蛋白,經轉氨作用可將D-谷氨酸轉變為D-丙氨酸(D-Ala),后者為細菌細胞壁肽聚糖的重要組成成分[11]。在單核細胞增生性李斯特菌中,存在兩種途徑生成D-Ala:基因dal編碼丙氨酸消旋酶(Alr),可將L-Ala轉化為D-Ala;基因dat編碼D-氨基酸氨基轉移酶蛋白,可將D-谷氨酸與丙酮酸通過轉氨作用生成D-Ala與α-酮戊二酸,當dat與dat基因全部缺失時,由于不能產生D-Ala,在無外源D-Ala添加時,該dat/dat雙基因缺失菌株將發生溶菌死亡[14]。當菌株缺失dat基因時,該缺失菌株在生長狀態、毒力基因表達水平、細胞侵襲性、生物被膜的生成量等方面的影響目前尚無研究報道。 作為一種潛在的疫苗載體,對李斯特菌進行減毒以保證安全性就顯得尤為重要。本研究以單增李斯特菌野生株EGDeactA及inlB雙基因缺失株(EGDe ΔactAΔinlB)為材料,利用同源重組的方法構建缺失了dat基因的菌株(EGDeΔactAΔinlBΔdat),并對構建的該缺失菌株的生長狀態、毒力基因表達水平、細胞侵襲性及生物被膜的生成量等方面做進一步分析。該項研究對菌株EGDeΔactAΔinlBΔdat的特性提供了參考,為研究dat基因的功能提供了材料。
1 材料與方法
1.1 材料
1.1.1 菌株和質粒 單增李斯特菌野生株EGDe、穿梭質粒pKSV7由上海交通大學史賢明教授贈予,EGDeΔactAΔinlB由本實驗室構建,大腸埃希菌(Escherichiacoli)DH5α感受態細胞購自天根生化科技(北京)有限公司。
1.1.2 主要試劑與儀器 膠回收試劑盒、質粒提取試劑盒,天根生化科技(北京)有限公司;PCR相關試劑、RT-PCR相關試劑、DNA連接酶、限制性內切酶,TaKaRa寶生物工程(大連)有限公司;腦心浸液培養基(BHI),北京陸橋技術有限公司;氨芐青霉素、氯霉素及其他常規試劑,國藥集團化學試劑有限公司;PCR熱循環儀、熒光實時定量PCR儀,Applied Biosystems公司;凝膠成像儀、Thermo濃度測定儀,美國Thermo基因有限公司;Mini-power電泳儀、電穿孔儀,美國伯樂公司;SpectraMax M2多功能酶標儀,美國分子儀器公司。
1.1.3 引物 根據GenBank登錄的基因序列(登錄號Gene ID:985722NC_003210.1),用Primer3 Input設計擴增上游同源臂引物P1/P2,下游同源臂引物P3/P4。連接后PCR引物為P1/P4,全長1 980 bp,缺失片段大小為839 bp。相關引物序列見表1,由生工生物工程(上海)股份有限公司合成。
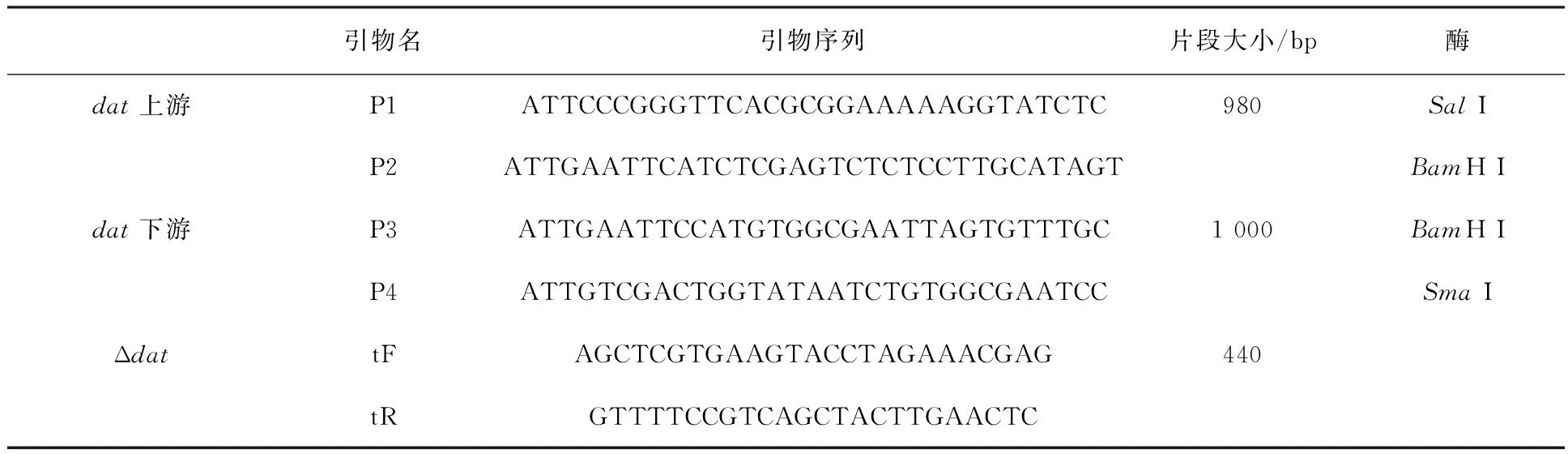
表1 PCR擴增所用的引物序列
1.2 方法
1.2.1 基因缺失株的構建 ①穿梭重組質粒的構建:引物P1/P2及P3/P4分別用于擴增dat基因的上下游同源臂,擴增片段分別經BamH I 單酶切后16 ℃過夜連接約16 h,以引物P1/P4對連接產物進行擴增,后經DNA回收試劑盒割膠純化回收,再經SalI與SmaI 酶切。酶切片段經瓊脂糖電泳后割膠純化回收,與經SalI與SmaI酶切的穿梭質粒pKSV7連接,并轉化至大腸埃希菌DH5α中,涂布含氨芐青霉素(100 μg/mL)的LB固體培養基培養。②電轉化與同源重組:引物P1/P4對LB平板上的單菌落進行PCR鑒定。鑒定為陽性的克隆抽提質粒后送華大基因測序。測序正確的重組質粒經12.5 kV/cm、3 ms電轉化至EGDe ΔactAΔinlB中,涂布含氯霉素(10 μg/mL)的抗性平板培養24~48 h,引物P1/P4對平板上的單菌落進行鑒定。鑒定為陽性的克隆在41 ℃和氯霉素雙重壓力下傳8代,然后在30 ℃無抗性條件下傳6代,末代培養物在無抗性BHI平板上劃線。③缺失株的鑒定:挑取BHI平板上的單菌落于液體培養基培養,分別用引物P1/P4及tF/tR進行PCR鑒定,鑒定成功的菌株分別劃線于BHI平板及含氯霉素(10 μg/mL)抗性平板,37 ℃培養。
1.2.2 缺失株生長曲線測定 利用酶標儀對EGDe ΔactAΔinlB及EGDeΔactAΔinlBΔdat的生長能力進行測定。挑取BHI平板上的單菌落37 ℃過夜培養后,按1∶100轉接到新鮮的液體培養基中,37 ℃搖床培養。每間隔1 h吸取樣品于96孔微孔板中,酶標儀測定600 nm處的吸光度光密度值(OD值)后繼續37 ℃搖床培養,連續測12 h。
1.2.3 毒力基因表達水平的檢測(RT-PCR) 為確定該基因對其他毒力基因及dal表達量的影響,本實驗比較了兩株菌在多個基因表達量上存在的差異。分別挑取EGDeΔactAΔinlB及EGDe ΔactAΔinlBΔdat平板上的單菌落過夜培養后,各取5 mL飽和菌液提取RNA后,按照TaKaRa反轉錄試劑盒的操作說明先去除其中的DNA,再反轉錄為cDNA。以cDNA為模板進行qRT-PCR,實驗中所用的其他毒力基因引物見表2。

表2 qRT-PCR所用的引物序列
1.2.4 生物被膜的形成量 構成生物被膜的胞外多糖可與結晶紫結合,可利用胞外多糖的量來衡量生物被膜的生成量。將過夜培養的細菌用新鮮的BHI液體培養基調整至OD600為0.15,取200 μL加入96孔細胞培養板中,37 ℃培養48 h。無菌PBS洗3遍后,1%的結晶紫染色30 min,無菌水洗3遍,每孔滴加95%乙醇洗脫30 min,利用酶標儀測量其在570 nm下的吸光度值。
1.2.5 細胞侵襲實驗 將生長良好的Caco-2細胞消化并轉移至12孔細胞培養板中,過夜培養。細菌侵襲前,以感染復數MOI=100∶1的比例加入新鮮的待測菌液共培養2 h。侵襲結束后,換用慶大霉素(200 μg/mL)37 ℃處理30 min。將慶大霉素吸出,無菌生理鹽水輕柔清洗2次,1% Triton X-100裂解細胞,每孔充分吹打并收集至離心管中,生理鹽水梯度稀釋涂布BHI平板,置于37 ℃培養箱培養并計數。
2 結果與分析
2.1 上下游片段擴增及連接
上下游同源臂片段經1.5%瓊脂糖凝膠電
泳,條帶大小分別在980 bp和1 000 bp左右(圖1a),連接后產物經電泳,顯示一條約1 980 bp的條帶(圖1b),與預期結果一致。
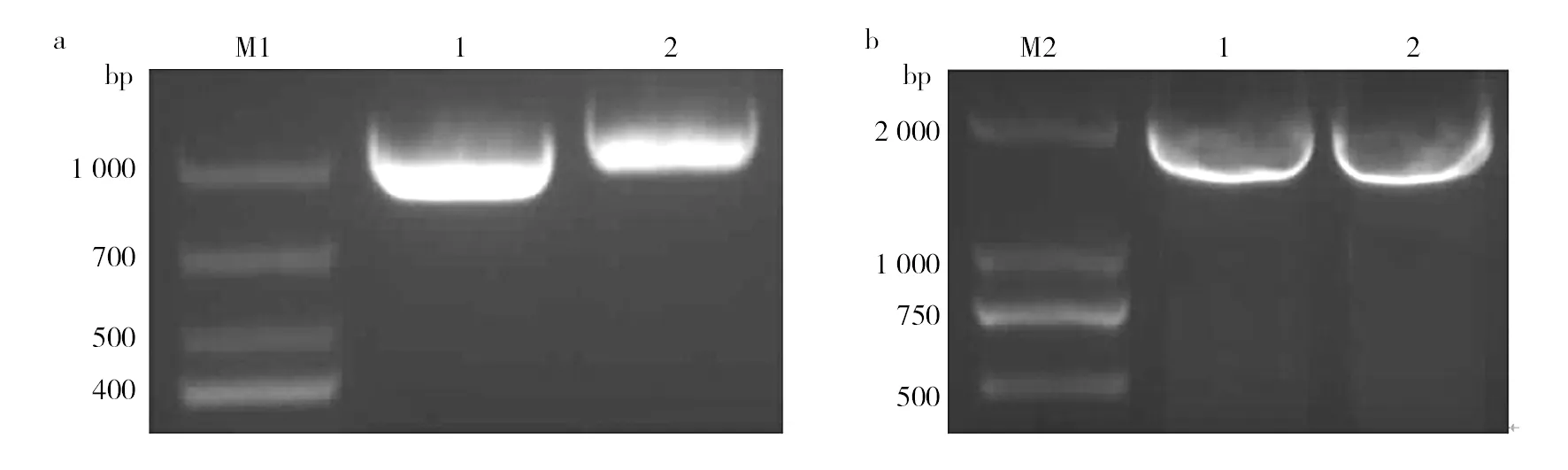
圖1 上下游同源臂及連接后PCR擴增結果Fig.1 The PCR amplification results of upstream and downstream homologous arm and after connecting a1:上游同源臂;a2:下游同源臂;b:上下游連接后擴增片段;M1:DL 1 000 bp marker;M2:DL 2 000 bp markera1: Upstream homologous arm; a2: Downstream homologous arm; b: Amplified fragment after upstream and downstream homologous arm connecting; M1: DL 1 000 bp marker; M2: DL 2 000 bp marker
2.2 穿梭重組載體構建
連接片段經SalI及SmaI酶切后,與同樣經酶切的穿梭質粒pKSV7連接,并轉化至大腸埃希菌DH5α中。陽性克隆抽提重組質粒測序成功后,經雙酶切鑒定,可見1 980 bp左右的片段,大小與預期一致(圖2a),表明重組質粒構建成功。重組質粒電轉化鑒定結果如圖2b,可見1 980 bp左右的條帶(泳道3、4),表明重組質粒成功電轉化至EGDeΔactAΔinlB中。
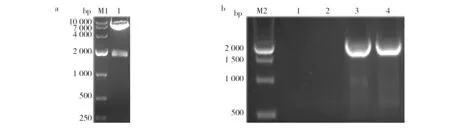
圖2 鑒定結果Fig.2 Identification results a:雙酶切鑒定結果; b:電轉化鑒定結果; M1:DL 10 000;M2:500 bp marker;b1~b4:單菌落a: Identification of double digestion; b: Identification of electrotransformation; M1: DL 10 000 bp marker; M2: 500 bp marker; b1-b4: Monoclonal strains
2.3 缺失株的鑒定
引物tF/tR的結合位點位于缺失的839 bp的序列上,若dat基因被敲除,則該引物將不能擴增出條帶。鑒定時,分別采用引物P1/P4及tF/tR對單菌落PCR擴增,若P1/P4擴增出1 980 bp左右的條帶,而tF/tR未擴增出條帶,則該菌株為疑似敲除菌株。如圖3a(1~2)所示,引物P1/P4擴增出1 980 bp左右的條帶;如圖3b(1~2)所示,引物tF/tR未擴增出條帶,表明1、2號為疑似敲除株,經氯霉素抗性進一步鑒定,該克隆不能在抗性平板上生長,確定為敲除株。
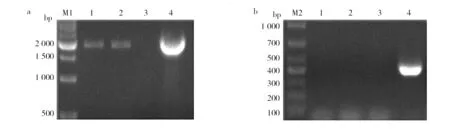
圖3 基因缺失株的鑒定Fig.3 Identification of gene deletion stain a:引物P1/P4擴增產物; b:引物tF/tR擴增產物; 1~2:單克隆菌株;3:陰性對照;4:陽性對照; M1:500 bp marker;M2:DL 1 000 markera: PCR products of P1/P4 primers; b: PCR products of dat-F/dat-R primers 1-2: monoclonal strains; 3: Negative control; 4: Positive control; M1: 500 bp marker; M2: DL 1 000 marker
2.4 生長曲線測定
酶標儀測定12個時間點的600 nm處的光密度值繪制生長曲線,如圖4所示,與親本株EGDeΔactAΔinlB相比,敲除株EGDeΔactAΔinlBΔdat培養5~8 h的菌濃度低于親本株(P<0.01),而在10 h后達到的菌濃度與親本株相同。表明dat基因的缺失使菌株通過另一途徑經丙氨酸消旋酶作用產生D-Ala,但在菌株快速增長的對數期,單一基因產生的D-ala不足以維持菌株生長需要,因而對數期菌濃度低于親本株。
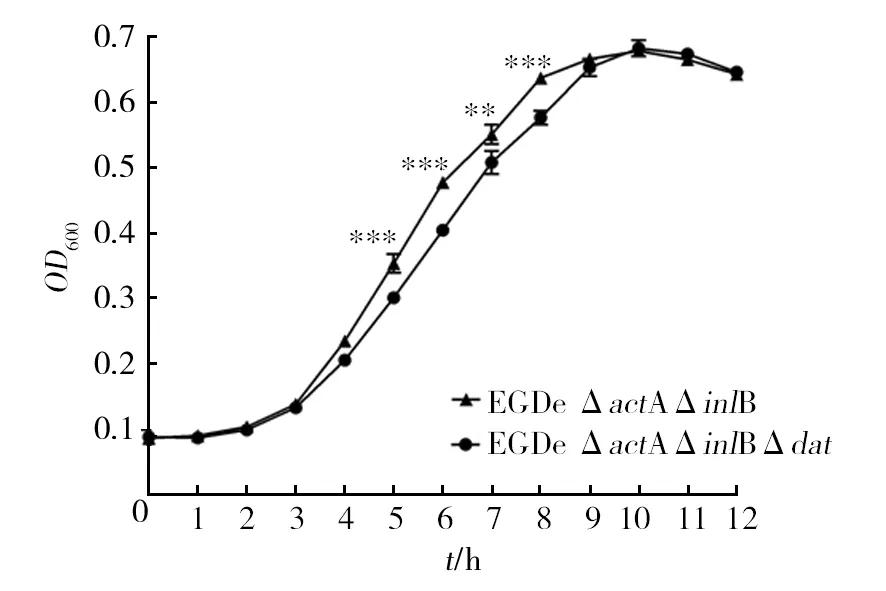
圖4 生長曲線Fig.4 Growth curve **:差異顯著(P<0.01);***:差異顯著(P<0.001),下圖同 **: Significant difference (P<0.01); ***: Significant difference (P<0.001),same the follow
2.5 毒力基因的相對表達量
本實驗選取了李斯特菌EGDe的12個毒力基因及與D-Ala合成有關的2個基因,比較EGDe ΔactAΔinlB與EGDe ΔactAΔinlBΔdat在基因表達量上存在的差異,其中以EGDe ΔactAΔinlB的基因表達水平為對照組,即EGDeΔactAΔinlB的每個目的基因表達水平均以1表示,EGDe ΔactAΔinlBΔdat的目的基因表達水平大于1為表達上調,反之則為表達下調。由圖5,可知菌株EGDe ΔactAΔinlBΔdat的dat基因無信號,進一步驗證dat基因的缺失;EGDe ΔactAΔinlBΔdat與EGDeΔactAΔinlB相比,基因srtA、plcA表達量下調(P<0.01),VIP、inlA基因表達上調(P<0.01)。
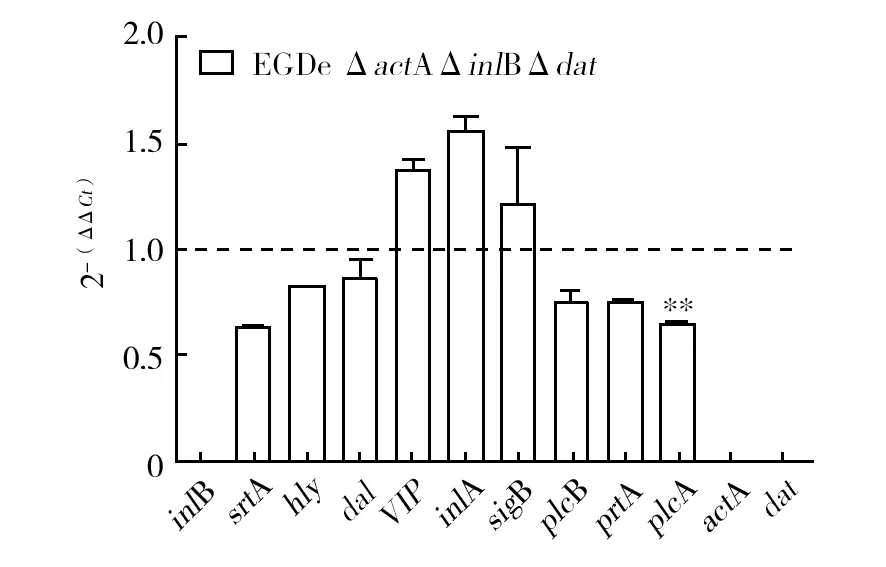
圖5 qRT-PCR結果Fig.5 Results of qRT-PCR
2.6 生物被膜的生成量
生物被膜是細菌抵抗外界不利環境時形成的,它是一種多細胞聚合物,由細菌團塊與表多糖、蛋白和胞外DNA構成的細胞外基質形成的復合物。由圖6可知,與親本株EGDe ΔactAΔinlB相比,dat基因的缺失使得菌株形成生物被膜的能力顯著增加(P<0.01),而在培養基中添加D-Ala的缺失株菌膜生成量與EGDe ΔactAΔinlB相比無顯著差異,推測由于dat基因的缺失使菌株處于D-Ala營養不足的脅迫狀態,因而對于外界不利條件形成自我保護的生物被膜。
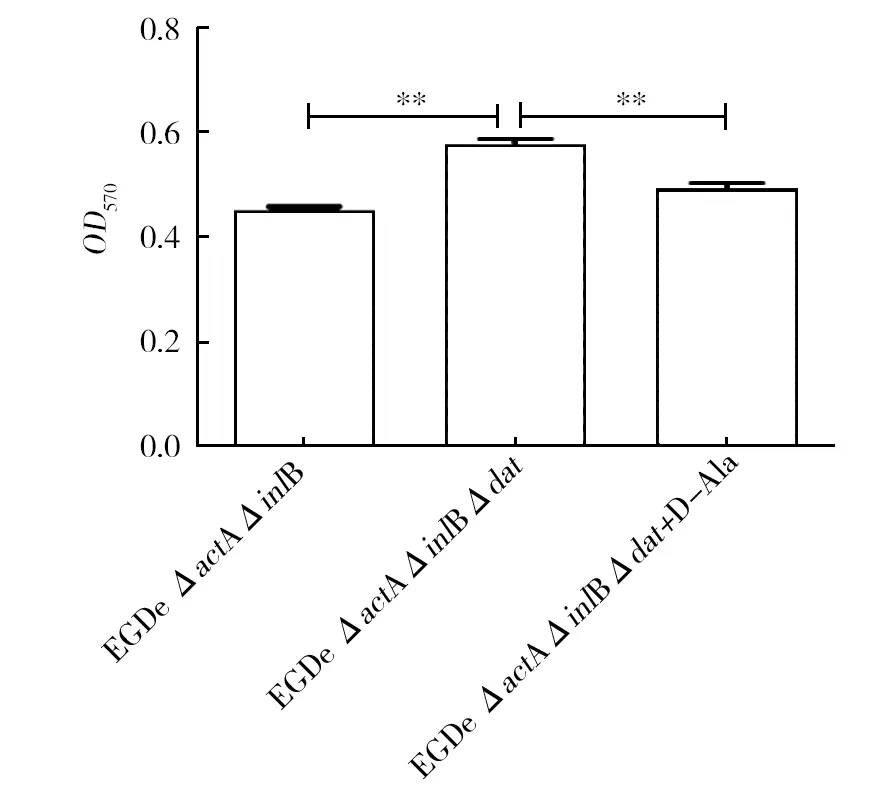
圖6 生物被膜生成量比較Fig.6 Comparation of biofilm
2.7 細胞侵襲結果
由Caco-2細胞侵襲結果(圖7)可知,敲除actA及inlB基因后,菌株EGDe ΔactAΔinlB的侵襲能力顯著下降(P<0.05),表明actA及inlB基因在細胞侵襲過程中發揮了重要作用。缺失dat基因,并未對侵襲結果產生顯著影響,表明dat基因敲除后,菌株的侵襲能力并無發生變化,也不會引起菌株毒力返祖的現象。
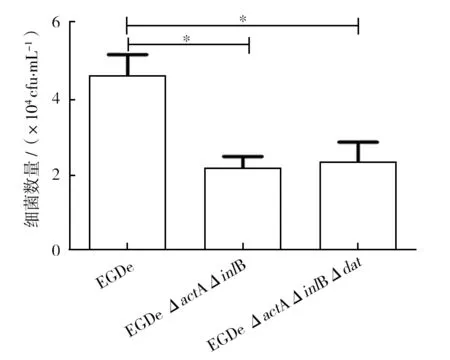
圖7 細胞侵襲結果Fig.7 Results of cell infection *:差異顯著(P<0.05) *: Significant difference (P<0.05)
3 討 論
作為一種胞內寄生的模式菌,闡明 Lm 致病的分子機制對于研制具有應用前景的弱毒疫苗和活疫苗載體具有十分重要的意義。相關報道表明,單增李斯特菌識別、黏附與侵入宿主細胞的過程與內化素基因(inl)編碼的內化素A和B有關[18-19],而其在細胞間運動及擴散由毒力基因plcA、plcB、hly、actA等介導[20-22]。根據文獻,單增李斯特菌dal/dat雙基因缺失株的毒力將明顯減弱,與野生株相比,對小鼠的半數致死率可提高4個數量級[23]。本研究在單增李斯特菌actA及inlB雙基因缺失株的基礎上,利用同源重組成功構建了dat基因缺失的三基因缺失株,并對該缺失菌株的生長特性狀態、毒力基因表達的變化、生物被膜的生成量及對細胞的侵襲性進行了研究。結果顯示,缺失dat基因的菌株在無外源D-Ala補充時雖能生存,但缺失株對數期的菌濃度顯著低于EGDe ΔactAΔinlB(P<0.01),可能由于dal基因產生的D-丙氨酸的量不足以維持菌株快速生長的需要,因而對數期的菌濃度低于親本株。RT-PCR結果顯示,與親本株EGDe ΔactAΔinlB相比,基因srtA、plcA表達量下調(P<0.01),VIP、inlA基因表達上調(P<0.01)。缺失株菌膜生成量顯著增加(P<0.01),培養基中添加D-Ala后菌膜生成量與親本株相比無差異,表明基因dat缺失使菌株處于一種外界營養脅迫狀態。缺失株對Caco-2細胞的侵襲與親本株相比無顯著差異。本研究表明基因dat在菌株生長及菌膜生成上起重要的調控作用,但不會影響菌株毒力。
在革蘭陽性菌,如鼠傷寒及豬霍亂沙門氏菌中[24],同樣存在類似功能的基因—asd基因,編碼天冬氨酸β-半乳糖脫氫酶。asd缺失菌株在無外源二氨基庚二酸存在的條件下會發生溶菌死亡,通過質粒回補asd基因并攜帶外源抗原基因而表達相應抗原蛋白,能有效解決質粒攜帶外源基因表達不穩定的問題,為沙門氏菌活疫苗載體的研發開辟了新的途徑。由于單增李斯特菌中存在兩個功能類似的基因dal與dat,而由本研究表明,單獨缺失dat基因并不能構建李斯特菌的營養缺陷體,的因而下一步計劃是在本研究的基礎上進一步缺失dal基因,通過質粒回補dal或dat基因并攜帶外源抗原基因,構建能穩定表達外源基因的單增李斯特菌活疫苗載體。該項研究通過對缺失株生長特性、基因的相對表達量、生物被膜的生成量及對細胞的侵襲性的研究,為基因dat缺失后菌株的生物學特性提供依據,并對進一步缺失dal基因,構建李斯特菌活疫苗載體提供參考。
猜你喜歡
藝術評鑒(2022年6期)2022-05-10 10:07:27
樂府新聲(2019年2期)2019-11-29 07:34:30
幼兒園(2019年7期)2019-09-05 17:49:18
北方音樂(2017年4期)2017-05-04 03:40:15
中國音樂教育(2015年8期)2015-05-16 03:22:06
小小說月刊(2013年6期)2013-05-14 14:55:19
食品科學(2013年23期)2013-03-11 18:30:07
食品科學(2013年23期)2013-03-11 18:30:05
食品科學(2013年15期)2013-03-11 18:25:40
食品科學(2013年15期)2013-03-11 18:25:25
