基于離子印跡聚合物微球固相萃取/微波等離子體發射光譜法測定地表水中痕量鉛
2018-09-06 10:51:26梁維新潘佳釧宋玉梅郭鵬然
分析測試學報 2018年8期
梁維新,潘佳釧,宋玉梅,郭鵬然
(廣東省測試分析研究所 廣東省水環境污染在線監測工程技術研究中心 廣東省化學危害應急檢測技術重點實驗室,廣東 廣州 510070)
地表水中的鉛主要來源于工業活動產生的廢水污染、含鉛燃料的廢氣沉降、重金屬礦的開采冶煉以及含鉛制品的生產使用等[1]。鉛可通過污染飲用水源以及食物鏈富集等途徑進入并在人體蓄積,導致人體神經、造血、骨骼以及消化等多個系統產生嚴重損傷[2-3],嚴重威脅人體健康。
目前鉛的檢測方法主要包括原子吸收光譜法(AAS)、電感耦合等離子體發射光譜法(ICP-AES)以及電感耦合等離子體質譜法(ICP-MS)等。其中AAS法需使用乙炔等易爆炸性氣體,安全隱患高[4];ICP-AES法檢出限高,且消耗大量價格昂貴的氬氣;ICP-MS法檢出限低、線性范圍寬,但儀器價格昂貴,且使用氬氣為工作氣體,運行成本高[5]。目前商品化的微波等離子體發射光譜儀(MP-AES)可直接采用空氣與氮氣運行,成本低,安全性高,線性范圍寬,且與ICP-OES的檢測性能相近,但其對鉛的檢出限比生活飲用水衛生標準(GB 5749-2006)以及地表水環境質量標準(GB 3838-2002)限值高,制約了其在水中鉛的分析上的應用。
離子印跡技術是一種通過合成對模板離子具有特異性識別能力的聚合物而實現萃取和目標離子富集的技術,將離子印跡技術結合固相萃取[6-8]、磁性固相微萃取[9-11]、電化學傳感器[12-14]以及膜分離[15]等技術實現鉛的富集和檢測已多有報道。與其他富集方法相比,固相萃取法具有成本低、選擇性高、富集因子高等優勢[16],目前已得到了廣泛應用,其富集材料大多采用本體聚合法制備,但需要破碎研磨步驟,且得到的聚合物形狀不規則。皮克林乳液聚合法通過水/油兩相界面的固體顆粒維持和穩定乳液結構[17],可減少對環境有害的表面活性劑的使用,且無需破碎研磨即可得到形狀規則的聚合物微球,方法簡便。本文通過皮克林乳液聚合法合成離子印跡微球,將鉛離子印跡固相萃取技術與MP-AES聯用建立了地表水中痕量鉛的測定方法,并應用于實際水樣的分析。
1 實驗部分
1.1 儀 器
微波等離子體原子發射光譜儀(Agilent 4100 MP-AES),儀器工作條件:常規霧化進樣系統(CNSI)霧化室;Concentirc nebulizer霧化器,霧化器壓力為240 kPa;積分時間3 s,樣品提升時間10 s,穩定時間10 s;3通道蠕動泵;背影校正Auto方式。三重四極桿電感耦合等離子體串聯質譜儀(Agilent 8800),儀器工作條件:射頻功率1.55 kW;霧化器:MicroMist型;等離子氣流量:15.0 L/min;載氣流量:0.9 L/min;輔助氣流量:0.9 L/min;采樣深度:8 mm;掃描方式:跳峰;氧化物比率:1.12%;雙電荷比率:1.56%;內標:115In。SHA-BA型恒溫水浴振蕩器(常州奧華儀器有限公司);BSA224S-CW型電子分析天平(賽多利斯公司);TDL-40B型離心機(上海安亭科學儀器廠);KQ-500E型超聲發生器(昆山市超聲儀器有限公司);BF-2000F型氮氣吹掃儀(八方世紀科技有限公司)。
1.2 試劑與材料
甲基丙烯酸(MAA)、二甲基丙烯酸乙二醇酯(EGDMA)、偶氮二異丁腈(AIBN)均購自北京市百靈威科技有限公司;聚乙二醇辛基苯基醚(TX-100)購自江蘇強盛功能化學有限公司;氣相二氧化硅購自德國贏創德固賽有限公司;硝酸鉛購自天津市光復精細化工研究所;甲苯、甲醇、硝酸購自廣州化學試劑廠;以上試劑均為分析純,實驗用水為去離子水(18.2 MΩ· cm)。
地表水樣品分別采自珠江廣州河段、肇慶市鼎湖山及廣州市新塘飲用水源地,經0.45 μm濾膜過濾去除雜質后,常溫保存備用。
1.3 鉛離子印跡聚合物微球的制備
在具塞玻璃小瓶中加入0.49 g的MAA、20 mg的硝酸鉛、1.51 mL的EGDMA 以及0.5 mL甲苯,混合超聲3 min制備油相;將20 mg氣相二氧化硅、20 mg的AIBN溶于10 mL 0.3%的TX-100溶液中,超聲5 min制備水相。將油相和水相混合制備穩定的皮克林乳液。將皮克林乳液通氮氣2 min后,將玻璃小瓶置于恒溫油浴鍋中,于70 ℃引發自由基聚合,聚合17 h后將所得固體沉淀用甲醇和高純水反復浸泡清洗以除去可溶性物質,再加入30 mL的10%氫氟酸浸泡6 h以除去微球表面的二氧化硅,最后以10%硝酸通過索氏提取法去除離子印跡微球中的Pb2+,清洗并干燥,即得IIPMs固體微粒,合成方法原理如圖1所示。
非離子印跡微球(NIPMs)的制備方法與IIPMs的制備方法類似,但在合成過程不加入硝酸鉛。
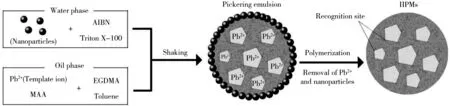
圖1 皮克林乳液聚合法合成IIPMsFig.1 Preparation of IIPMs via Pickering emulsion polymerization
1.4 特異性吸附性能的評價
離子印跡聚合物的選擇性吸附性能采用分配系數(Kd)、選擇性系數(k)、相對選擇性系數(k’)描述,各參數按以下公式進行計算[18]:Kd=qe/Ce;k=Kd(Pb)/Kd(x);k’=kIIPMs/kNIPMs。式中:qe為各離子的平衡吸附量(mg/g);Ce為各離子的平衡質量濃度(mg/L);Kd(Pb)為Pb2+的分配系數;Kd(x)為競爭離子的分配系數;kIIPMs為IIPMs的選擇性系數;kNIPMs為NIPMs的選擇性系數。
1.5 等溫吸附實驗
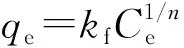
1.6 鉛離子印跡聚合物的固相萃取
準確稱取75 mg的IIPMs,將其填入1 mL的SPE空柱管中,上下放置篩板以防止微球泄漏,壓緊篩板后即得鉛離子印跡固相萃取柱。用高純水對固相萃取柱洗滌活化后,再將一定體積的Pb2+溶液過柱,通過外加負壓將柱抽至近干,用5%的HNO3溶液洗脫吸附在柱中的Pb2+,收集洗脫液,利用MP-AES法測定洗脫液中Pb2+含量,并計算加標回收率。
2 結果與討論
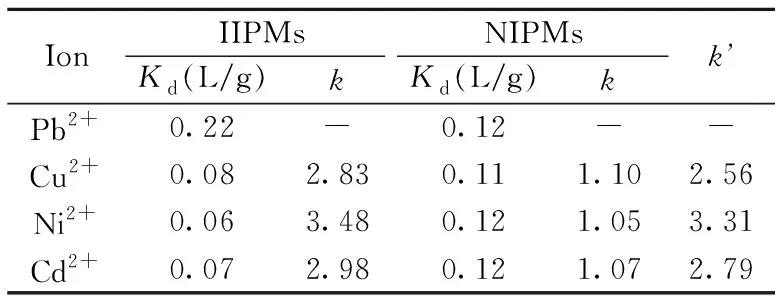
表1 IIPMs/NIPMs選擇性吸附評價結果Table 1 Selective adsorption parameters of IIPMs and NIPMs
2.1 特異性吸附性能的評價
本實驗選擇與Pb2+有相同離子電荷和不同離子半徑的Cu2+、Ni2+、Cd2+作為競爭吸附離子(半徑大小Pb2+=120 pm、Cd2+=114 pm、Ni2+=69 pm、Cu2+=71 pm)[19]評價IIPMs的選擇性吸附能力(表1)。結果顯示,IIPMs對Pb2+的分配系數(Kd)明顯高于競爭離子,是競爭吸附離子的2.8~3.5倍,表明IIPMs具有較好的特異性識別以及抗干擾能力;而相對選擇性系數(k’)均在2.5以上,證明了離子印跡微球對Pb2+具有很好的選擇吸附效果。
2.2 IIPMs固相萃取柱性能的評價
2.2.1吸附容量為考察吸附容量,分別將30、40、50、60、70 mL的Pb2+溶液(1.5 mg/L,pH 6.5)以0.5 mL/min的流速通過固相萃取柱,經4 mL 5%的HNO3洗脫并測定,結果表明其吸附回收率隨Pb2+加入量的增大而逐漸下降,當溶液體積低于50 mL(Pb2+的加入量≤75 μg)時,吸附回收率高于90%。若以90%吸附回收率進行評估,75 mg 填料量計算,則IIPMs對Pb2+的最大吸附量為1.0 mg/g,此結果與等溫吸附結果接近(見表2中qm)。同時考察了在高富集流速下IIPMs固相萃取柱對Pb2+的吸附情況,分別將20、40、60、80、100 mL的Pb2+溶液(20 μg/L,pH 6.5)以5 mL/min的流速通過固相萃取柱,實驗結果顯示,當樣品加入量在60 mL以內(Pb2+的加入量≤1.2 μg),以4 mL 5%的HNO3洗脫時,回收率均高于90%,故后續實驗控制Pb2+的加入量在1.2 μg以下,為兼顧最大富集倍數和儀器進樣量要求,后續實驗選擇洗脫體積為4 mL。

表2 IIPMs/NIPMs對Pb2+的等溫吸附擬合結果Table 2 Fitting results of isothermal adsorption of Pb2+adsorbed by IIPMs and NIPMs
2.2.2溶液pH值對萃取性能的影響采用IIPMs固相萃取柱以5 mL/min的流速分別對25 mL 20 μg/L的Pb2+溶液進行富集,然后用4 mL 5%HNO3洗脫,考察溶液pH值對富集效果的影響,結果表明,當溶液的pH值為3.0、4.0、5.0、6.0、7.0時,Pb2+的回收率分別為86.1%、91.9%、93.6%、104%和104%。這是由于低pH值溶液中大量的H+與Pb2+產生競爭吸附作用[20]而降低了富集效果,因此,IIPMs固相萃取柱在弱酸性或中性溶液中對Pb2+的富集效果最佳。
2.2.3溶液濃度對回收率的影響采用IIPMs固相萃取柱以5 mL/min的流速對25 mL不同質量濃度的Pb2+溶液進行富集,然后用4 mL 5%HNO3洗脫,以考察Pb2+濃度對富集效果的影響。結果顯示,當Pb2+的質量濃度為10、20、30、40 μg/L時,回收率分別為94.4%、101%、99.9%和101%,說明IIPMs固相萃取柱對不同濃度的Pb2+均具有良好的吸附效果。
2.2.4富集倍數將一系列1 μg/L的Pb2+溶液(pH 6.5)以5 mL/min的流速通過固相萃取柱,當溶液體積在1 000 mL以內時,吸附回收率高于90%,繼續增大溶液體積,吸附回收率則出現明顯下降,因此以4 mL洗脫液計算,IIPMs的最大富集倍數為250倍。由于富集溶液體積較大會導致富集時間過長,以最低富集溶液質量濃度(1 μg/L)計算,當富集倍數為25倍時,已達到MP-AES的定量下限20 μg/L(以5%HNO3為空白樣品,MP-AES連續測定11次,按10倍標準偏差計算),因此后續實驗選擇富集倍數為25倍。
2.2.5重復利用性能按照固相萃取程序對同一根IIPMs固相萃取柱進行吸附和洗脫操作,經重復使用12次后,IIPMs固相萃取柱對Pb2+仍具有良好的吸附和洗脫能力,加標回收率均在93%以上,表明IIPMs固相萃取柱的重復利用性能良好。
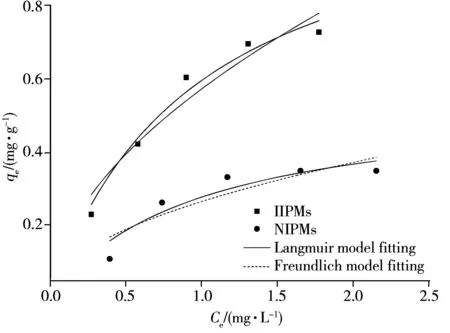
圖2 等溫吸附擬合曲線Fig.2 Curves of isothermal adsorption fitting
2.3 等溫吸附及形貌表征
按照“1.5”方法進行等溫吸附試驗,據表2和圖2可知,IIPMs吸附Pb2+的過程符合Langmuir等溫吸附模型,擬合優度(r2=0.969 7),屬于單分子層吸附,通過擬合計算得到最大單層飽和吸附容量為1.20 mg/L。IIPMs對Pb2+的吸附能力均明顯強于NIPMs,這可能是由于在合成過程中引入了鉛離子的IIPMs較NIPMs具有更小粒徑(IIPMs=26.6 μm,NIPMs=35.2 μm)以及更多孔的表面(圖3A、B),有利于增大微球的比表面積,從而提升其吸附能力。
2.4 線性范圍、方法精密度及檢出限
以5%HNO3配制一系列不同質量濃度的Pb2+標準溶液并繪制標準曲線,結果顯示,Pb2+在0.02~1 mg/L范圍內線性良好,相關系數(r)為0.999 6。在最優固相萃取條件下(富集體積100 mL,洗脫體積4 mL,pH 6.5),以1 μg/L和10 μg/L去離子水介質的Pb2+為富集溶液,按照固相萃取程序平行實驗8次并進行測定,相對標準偏差(RSD)分別為5.2%和2.4%,表明該方法具有良好的精密度。由于MP-AES對Pb2+的動態線性范圍可達4~5個數量級,洗脫后高、低濃度的樣品均可直接進樣分析。以去離子水為樣品,按照3倍空白標準偏差(σ)所對應的質量濃度以及富集倍數計算得方法的檢出限為0.26 μg/L,其靈敏度可滿足生活飲用水衛生標準(GB 5749-2006)以及地表水環境質量標準(GB 3838-2002)的測定要求。

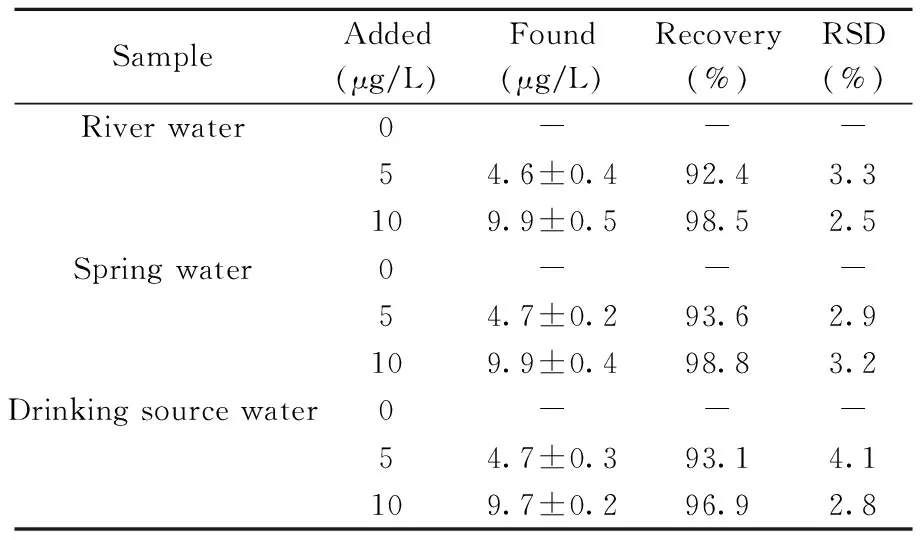
SampleAdded(μg/L)Found(μg/L)Recovery(%)RSD(%)River water0---54.6±0.492.43.3109.9±0.598.52.5Spring water0---54.7±0.293.62.9109.9±0.498.83.2Drinking source water0---54.7±0.393.14.1109.7±0.296.92.8
-:no detected
2.5 實際地表水樣的測定
在最優固相萃取條件下(富集溶液體積100 mL,洗脫溶液體積4 mL),采用本方法與直接進樣ICP-MS法對實際水樣進行測定,兩種方法在地表水樣品中均未檢出Pb2+;采用本方法測定地表水樣品的加標回收率為92.4%~98.8%,相對標準偏差不大于4.1%(表3)。表明該方法具有良好的準確度和精密度,可用于地表水中痕量鉛的檢測。本研究豐富了MP-AES技術的應用范圍。
3 結 論
本文以皮克林乳液聚合法制備了鉛離子印跡微球(IIPMs),并將鉛離子印跡固相萃取技術與微波等離子體發射光譜法聯用,建立了地表水中痕量Pb2+的離子印跡固相萃取/微波等離子體發射光譜測定方法。樣品富集后以5%HNO3洗脫,可直接進行MP-AES分析測定。固相萃取的最大富集倍數為250倍,富集時間為30~40 min。該方法的成本低,精密度良好,檢出限低,滿足生活飲用水衛生標準及地表水環境質量標準的要求。
