不同酶處理對生物解離乳狀液結構及穩定性的影響
2018-09-17 06:49:40齊寶坤謝鳳英鐘明明
農業機械學報 2018年9期
關鍵詞:界面
李 楊 李 紅 齊寶坤 謝鳳英 鐘明明 檀 政
(東北農業大學食品學院, 哈爾濱 150030)
0 引言
傳統的大豆油脂制取方式主要有機械壓榨法和溶劑浸出法。隨著研究的深入,人們發現可以利用生物酶法(水酶法)提取大豆油脂,該方法是一種綠色環保的新技術。在提取過程中,含有酶制劑的水充當油料種子的提取媒介,在最適的酶解條件下反應,酶解后離心分離得到游離油、乳狀液、水解液和殘渣4部分[1]。由于處理時間過長,且乳狀液難以分離,造成油脂無法被充分破除釋放,因而限制了油脂提取率[2]。因此,如何采用高效、綠色、安全的破乳方法將乳狀液中的油脂完全釋放是提高生物解離油脂提取率的重要環節。
乳狀液中加入酶能夠將油脂分離出來,主要是由于加入到乳液的酶能水解界面蛋白和表面磷脂,從而減小其分子大小,減小油滴界面的剛性,進而釋放被蛋白包裹的油脂。這些酶解反應可使油滴聚集變大,使得游離油更易釋放[3-5]。JUNG等[6]研究報道,乳狀液中加入LysoMax(磷脂酶A2)后破乳效果不好,這可能是由于形成了具有更強乳化性的溶血磷脂。磷脂酶D是大豆的一種內生磷脂酶,能將磷脂酰膽堿及磷脂酰乙醇胺兩種大豆磷脂轉變成磷脂酸。YAO等[7]研究表明,水酶法提取大豆油脂過程中,磷脂酰乙醇胺和磷脂酰膽堿向磷脂酸的轉變與油脂游離油得率的降低有一定的相關性。
因此,本文采用2種蛋白酶和3種磷脂酶對乳狀液進行處理,通過對粒徑分布、Zate電位、顯微鏡觀察、紅外光譜以及熒光光譜的測定,探究不同生物酶處理大豆乳狀液的蛋白質結構與穩定性之間的關系,明確乳狀液體系中關鍵組分和空間構象以及組分相互作用對乳化相形成及穩定的影響機制。
1 材料與設備
1.1 材料與試劑
大豆,東北農業大學大豆研究所;堿性蛋白酶Protex 6L,杰能科生物工程有限公司;十二烷基硫酸鈉(SDS)、β-巰基乙醇、磷酸氫二鈉、磷酸二氫鈉鹽酸、考馬斯亮藍(G-250)、牛血清蛋白(BSA)、氫氧化鈉、丙酮、無水乙醇等化學試劑均為分析純級。
1.2 儀器與設備
FW100型高速萬能粉碎機,紹興科宏儀器有限公司;擠壓膨化機,東北農業大學自制;BX53型正置生物顯微鏡,OLYMPUS公司;LGJ-1型冷凍干燥機,上海醫用離心機廠;GL-21M型高速冷凍離心機,上海市離心機械研究所; TNZ1-5700型傅里葉紅外光譜儀,英國Thermo Fisher公司;F2000型熒光光譜儀,日本Hitachi公司;Mastersizer 2000型粒度儀,英國馬爾文儀器有限公司。
2 試驗方法
2.1 生物解離大豆乳狀液的制備
參照齊寶坤[8]的方法,并作適當的修改,大豆粉碎后擠壓膨化預處理,得到膨化大豆粉。膨化大豆粉與水混合(液料比6 mL/g),加入堿性蛋白酶Protex 6L,攪拌均勻后在60℃水浴鍋中保持恒溫加熱,用2 mol/L NaOH調節pH值至9.0,酶解3 h后,取出于100℃沸水中滅酶10 min,在4 500 r/min轉速下離心20 min,離心后吸取游離油,將其余液體部分倒入分液漏斗,靜置24 h分層,將乳狀液分離。
2.2 乳狀液的生物酶處理
分別采用蛋白酶和磷脂酶處理乳狀液[9-10],每種酶在其最適的處理條件,酶解反應時間均為1 h,酶的添加量為2%(酶與乳狀液質量百分比)。堿性蛋白酶Protex 6L和堿性蛋白酶Acalase 2.4L在pH值為8、溫度為40℃條件下進行酶解;溶血磷脂酶在pH值為4.5、溫度為40℃進行酶解;磷脂酶A2和磷脂酶D在pH值為8、溫度為40℃條件下進行酶解,酶解后在3 585g下離心10 min,來分離乳狀液和水相。
2.3 乳狀液分層系數和游離油得率的測定
分層系數(Creaming index)的測定:取8 mL處理后的乳狀液于離心管中,在4℃貯藏24 h,乳狀液發生分層現象,上相渾濁,下相澄清。分層系數計算公式為
式中Hs——下相的高度
Hr——乳狀液總高度
游離油得率計算公式為
式中m——破乳后總游離油質量
M——乳狀液中含油質量
2.4 乳狀液粒徑及Zate電位的測定
采用激光光散射粒度分析儀測定乳狀液體積加權平均值(D4,3),將小部分乳狀液分散在50 mL蒸餾水中,保持粒徑數值在測定范圍內,大豆油滴的折射指數為1.47,分散劑的折射指數為1.333。測定在室溫(20℃)下進行,吸光度為0.001。
采用Zetasizer Nanozs 90型電位儀測定乳狀液溶液的Zate電位。0.05 mol/L pH值7.0的磷酸鹽緩沖溶液將大豆乳狀液樣品稀釋至質量分數為0.2%的溶液后進行測定,上樣體積1 mL,測定溫度25℃。每個樣品重復測量3次。
2.5 乳狀液正置顯微鏡分析
利用配有DP27型顯微數碼相機的BX53型正置生物顯微鏡對乳狀液的顯微結構進行觀察。用吸管取2~3滴乳狀液于干凈干燥的載玻片上,輕輕地加上蓋玻片后,置于顯微鏡明場放大100倍進行觀察。
2.6 乳狀液中蛋白質的提取
乳狀液水相中蛋白質提取方法采用丙酮沉淀法[11],將20 g/mL冰丙酮在-18℃下反應2 h,離心分離(12 000g,15 min,4℃),除去上清液,將沉淀繼續用冰丙酮洗滌4~5次,直到溶劑由黃色變成無色,沉淀中溶劑揮發后凍干得到蛋白,放入4℃冰箱備用。
2.7 表面疏水性的測定
依據LAEMMLI[12]的方法,即ANS(8-苯氨基-1-萘磺酸銨鹽)熒光探針法。稱取0.025 g不同處理方式下的蛋白樣品溶于50 mL磷酸鹽緩沖液中,配成pH值為7、0.01 mol/L的溶液,將溶液在室溫下攪拌混合1 h,然后在10 000g下離心處理30 min,取上清液采用Lowry法測定其蛋白質濃度,并用上述磷酸鹽緩沖溶液依次稀釋后,使其濃度在0.005~0.5 mol/mL之間,取不同濃度樣品的溶液4 mL,分別加入40 μL濃度為8 mmol/L的ANS溶液,經振蕩混合后靜置5 min,再測定樣品的熒光強度。試驗中激發波長為370 nm,發射波長為490 nm,夾縫寬為5 nm。
2.8 紅外光譜的測定
將樣品粉碎后過100目的篩子,粉末在40℃干燥箱中干燥12 h,然后在紅外燈下進行研磨處理。稱取約2 mg的待測樣品,加入200 mg溴化鉀,在瑪瑙研缽中研磨15 min,隨后進行壓片處理,壓片機在140 N壓力下約保持1 min,然后將制得的均勻透明薄片放入紅外光譜儀中進行測定。測定的條件為:掃描范圍400~4 000 cm-1,分辨率為4 cm-1,掃描信號累加64次。每種處理的圖譜掃描重復3次。
譜圖的分析處理采用Peakfit軟件,在酰胺Ⅰ帶1 600~1 700 cm-1進行兩點基線校正,然后再采用Savitsk-Golay函數進行平滑處理,求二階導數譜,并采用Gauss峰形進行擬合,然后估算出子峰的個數與位置,經過手動調整各子峰的峰高和半峰寬,進行多次擬合使得殘差最小,確定各子峰與各二級結構的對應關系后,根據其積分面積就可以計算出4種二級結構的相對含量。
2.9 熒光光譜的測定
依據尹壽偉[13]的方法,采用F-4500型熒光分光光度計測定乳狀液分離出的大豆蛋白色氨酸熒光光譜。將大豆蛋白樣品分別分散于0.01 mol/L、pH值為7.0的磷酸緩沖液中,配制蛋白質量濃度為0.15 mg/mL的溶液。熒光發散光譜分析以蛋白質分子內部的色氨酸熒光基團為探針,為了降低酪氨酸的貢獻,熒光光譜的激發波長為290 nm,光譜的掃描范圍為300~400 nm,激發與發射狹縫的寬均為5 nm。
2.10 乳狀液中磷脂的提取與測定
乳狀液中的磷脂提取方法如下[14]:
(1)粗油提取:向乳狀液中加入200 mL甲醇,混合搖勻后加入400 mL氯仿繼續混勻后,在室溫下磁力攪拌4 h,然后采用布式漏斗回收固體和萃取液,將固體反復萃取,將得到的萃取液合并,萃取液經過旋轉蒸發除去溶劑,繼續用氯仿、甲醇、0.74%水溶性KCl按照體積比8∶4∶3進行洗滌,最終的粗油儲存在-26℃下備用。
(2)磷脂濃縮:將100 mL正己烷與100 mL體積分數為87%的乙醇混合,混合后放入分液漏斗中,使之靜置平衡,靜置后分離得到上層正己烷相(溶劑A)和較低的乙醇相(溶劑B);約10 g粗油在200 mL分液漏斗中與45 mL溶劑A和15 mL溶劑B混合,5 min后相達到平衡,收集較低的乙醇相,15 mL溶劑B加入分液漏斗上層相中,相平衡后將較低的乙醇相收集與第1次得到的乙醇相合并,此過程重復10次,乙醇提取物與130 mL氯仿和0.1 mol/L K-EDTA(111 mL,pH值7)混合,然后收集較低的氯仿相,采用硫酸鈉干燥,除去溶劑的濃縮油在-26℃儲存待用。
乳狀液磷脂測定采用31P-NMR(磷-31核磁共振)分析,濃縮的粗油(80~90 mg)加入TPP(磷酸三苯酯,10 mg,固體)溶解在氯仿、甲醇Cs-EDTA組成混合液中,劇烈震蕩搖勻,離心后將較低的部分轉移到5 mm NMR瓶中進行測定。31P-NMR測定條件如下:探頭溫度29℃,脈沖寬度22 μs,掃描寬度9 718 Hz,采集時間1、2 s,間隔10 s,掃描次數256次。
3 結果與討論
3.1 分層系數和游離油得率
分別采用堿性蛋白酶Protex 6L、堿性蛋白酶Acalase 2.4L、溶血磷脂酶、磷脂酶A2和磷脂酶D處理的乳狀液,乳狀液的分層系數和游離油得率如圖1所示,堿性蛋白酶Protex 6L、堿性蛋白酶Acalase 2.4L分別在pH值為8的條件下進行水解,與原始乳液相比,其分層系數和游離油得率顯著增加,游離油得率分別達到90.05%和94.24%,油脂得率的增加可能是由于采用蛋白酶處理乳狀液時,蛋白酶能水解蛋白生成短肽,進而破壞了乳狀液界面的完整性,使得其不再具有足夠力量阻止油脂的聚集,油脂得以釋放[15-16]。采用磷脂酶處理后游離油得率均增加,有研究表明磷脂酶處理水解包裹在油脂表面的磷脂,降低乳狀液的穩定性,進而釋放游離油[17]。溶血磷脂酶處理效果較好,游離油得率都可達到95.12%左右,可能由于溶血磷脂酶處理的pH值為4.5,采用磷脂酶D和磷脂酶A2處理的pH值為8,磷脂酶A2處理后游離油得率較前兩種酶有所降低,為86.67%左右。本研究說明大豆蛋白和磷脂在液滴的界面上對乳狀液的穩定性起著很重要作用,這與JUNG等[18]研究結果相一致。
3.2 乳狀液穩定性
圖2和圖3所示為不同種類的生物酶對形成乳狀液處理后的Zeta電位和粒徑分布的影響。原始乳狀液的Zeta電位值為(-21.57±1.88) mV,經兩種堿性蛋白酶Protex 6L和Acalase 2.4L處理后乳狀液的負電位絕對值降低,乳狀液粒徑分布主要集中在50~100 μm,可能的原因是經由蛋白酶水解后使水解物分子量變小并水解成小分子的多肽[19],因而會導致在油滴表面的吸附量降低,再重新與磷脂復合吸附在油-水界面,所形成的膜強度就會下降;另外一方面,蛋白的過度水解使表面電荷減少、空間位阻的作用下降,也會造成了大量油滴的聚集,使乳液的粒徑增大[20]。磷脂酶A2酶解的乳狀液負電位絕對值較大,粒徑較小,分布范圍較窄,大粒徑液滴數量減少,乳化體系相對較為穩定,說明此時表面所帶的負電荷較多,乳狀液中磷脂非極性基團數量減少,親水性基團增強,使表面的蛋白與酶解后的磷脂相互作用增強,較好地吸附在油水界面,進而使乳液的粒徑變小[21],這與CHEN等[22]的研究結果相類似。經溶血磷脂酶酶解后其電位值為(-4.8±0.6) mV,出現較大部分粒徑的分布,而經磷脂酶D酶解的乳狀液電位變化到(-11.8±0.8) mV,這可能是由于磷脂酶D作用后使磷脂的結構發生改變甚至被破壞,使乳狀液表面所帶電荷發生變化,但此時表面電荷密度不大,液滴之間的靜電斥力也相對較小[23],溶血磷脂酶和磷脂酶D主要作用磷脂的疏水尾部[24],由于被蛋白包裹乳液界面的磷脂被水解的程度相對較弱,但可能會造成磷脂結構略微不穩定,形成了較大的乳滴。
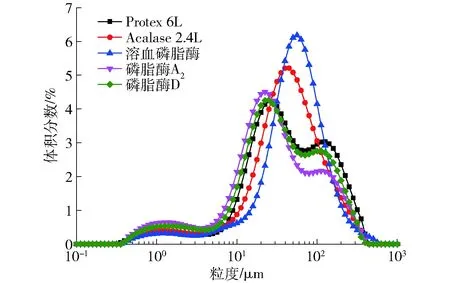
圖3 不同酶處理對乳狀液粒徑分布的影響Fig.3 Effect of different enzymes treatments on particle size distribution of oil emulsion
3.3 乳狀液微觀結構
不同酶處理后乳液的微觀結構采用光學顯微鏡進行觀察,如圖4所示。不同酶進行酶解處理乳狀液呈現不同的形態。從圖4可看出,經Protex 6L和Acalase 2.4L兩種堿性蛋白酶處理后的乳狀液微觀結構相似,出現大尺度油滴的聚集;溶血磷脂酶酶解后的體系乳狀液液滴中出現了油滴的成片聚集,乳狀液界面出現了破裂,且液滴形狀不規則、大小不均一,溶血磷脂酶水解后乳狀液界面上磷脂被水解,界面強度下降,乳液極不穩定,隨著油滴的聚集,碰撞使油滴之間出現聚結,從而使液滴粒徑進一步增大,導致出現有較多的大油滴,此外,在pH值為4.5的條件下,乳狀液中油滴也會相互聚集。磷脂酶D與堿性蛋白酶處理后的乳液分布狀態相似,油滴的聚集和粒徑的分布介于中間,磷脂酶A2酶解后的體系乳狀液系中乳滴較小,可能是由于乳液界面磷脂部分水解形成的水解產物與蛋白重新乳化后表面帶有更多電荷,具有抵抗乳液液滴聚集的能力[25],說明磷脂酶A2對乳狀液破除效果不好。由光學顯微鏡觀察來看,酶解后乳狀液粒徑、油滴的分布大小差別較大,表明油滴之間發生聚集現象,其乳狀液形態與粒徑大小測定結果一致。

圖4 不同酶酶解對乳狀液微觀結構的影響Fig.4 Effects of different enzymes treatment on microscopic structure of emulsion
3.4 乳狀液中蛋白質分析
3.4.1乳狀液表面疏水性
經不同酶酶解后乳狀液界面蛋白的表面疏水性指數是用來表征酶解后蛋白表面疏水基團數量的一個重要指數,也與蛋白的乳化特性有關,蛋白質的表面疏水性指數越高,其乳化能力越強[26]。圖5為不同酶酶解后乳狀液界面蛋白的表面疏水性變化,乳狀液界面的磷脂和蛋白在酶作用下,會影響界面蛋白的表面疏水性,從而影響相互作用及界面強度。經Protex 6L和Acalase 2.4L兩種堿性蛋白酶處理后的乳狀液界面蛋白表面疏水性與原始乳液相比都呈現下降,主要是由于蛋白酶會影響界面吸附蛋白的分子結構,導致游離的疏水基團裂解或者暴露出的疏水基團發生聚集,引起水解后蛋白表面疏水性的降低。其中Acalase 2.4L處理的表面疏水性最小,說明此時蛋白水解物中有更多的親水基團暴露,另外蛋白疏水性的變化會對蛋白的乳化性有重要影響,蛋白酶的過度酶解使得界面維持蛋白的內部結構力(包括氫鍵、范德華力、離子鍵等)逐漸被破壞,使油滴表面的保護層越來越薄,導致了乳化性的降低[27]。而用磷脂酶A2和磷脂酶D處理后的蛋白表面疏水性降低不明顯,可能是由于磷脂經酶解后頭部結構被破壞[28],但疏水的尾部并沒有暴露,與蛋白相互作用時,被緊密包埋在區域內,導致疏水性沒有明顯增加。而采用溶血磷脂酶處理的乳狀液,由于pH值的作用,其表面疏水性明顯降低。
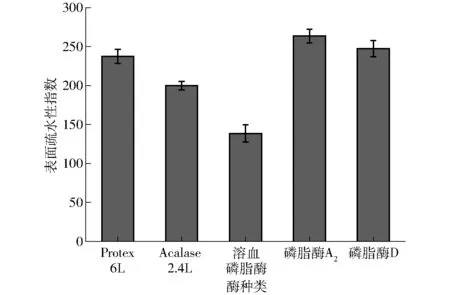
圖5 不同酶酶解后乳狀液蛋白的表面疏水性Fig.5 Surface hydrophobicity of oil emulsion with different enzyme treatments
3.4.2乳狀液中蛋白質紅外光譜分析
圖6 不同酶處理條件下乳狀液中蛋白質的紅外光譜圖Fig.6 FTIR of protein in oil emulsion with different enzymes treatment
乳狀液中的蛋白二級結構組成與蛋白分子的空間構象密切相關,因而會影響蛋白的功能特性。紅外光譜可顯示蛋白質酰胺Ⅰ帶、酰胺Ⅱ帶、酰胺Ⅲ帶等蛋白質結構中的相關信息[29]。傅里葉變換紅外光譜的研究可以定量給出每組水解物中蛋白質的二級結構含量。圖6為不同酶處理條件下乳狀液中蛋白質的紅外光譜圖。研究表明,蛋白質α-螺旋二級結構對應波數為1 646~1 664 cm-1;β-折疊結構對應波數為1 615~1 637 cm-1和1 682~1 700 cm-1;β-轉角結構對應波數為1 664~1 681 cm-1;無規卷曲結構對應波數為1 637~1 645 cm-1[30]。采用Gauss面積法擬合,通過峰位歸屬確定不同酶解物的二級結構種類和含量,計算結果見表1。原始乳狀液的二級結構組成為:α-螺旋相對含量(17.30±0.10)%,β-折疊相對含量(40.90±0.18)%,β-轉角相對含量(18.40±0.07)%,無規卷曲相對含量(23.40±0.12)%。由表1可知,其中酶解后的乳狀液中蛋白的二級結構主要以β-折疊和無規卷曲為主,Protex 6L和Acalase 2.4L堿性蛋白酶酶解后,蛋白質中α-螺旋相對含量降低,分別降到(14.58±1.18)%和(10.42±1.28)%,無規卷曲結構相對含量升高。這可能是由于α-螺旋和β-折疊屬于相對規則的構象,而β-轉角、無規卷曲的蛋白質分子結構比較疏松,經酶解后蛋白質的肽鏈變得伸展,柔韌性增加,疏水性基團暴露,致使在乳狀液中蛋白質二級結構相對含量不同,有利于吸附在油滴表面[31]。采用溶血磷脂酶、磷脂酶A2和磷脂酶D處理后的蛋白質α-螺旋相對含量均降低,無規卷曲相對含量升高,所發生的變化可能是由于酶解的磷脂水解物與蛋白分子發生交互作用時,部分影響了二級結構的轉移。其中溶血磷脂酶的無規卷曲相對含量達到(32.40±0.15)%,反應pH值4.5使得蛋白發生變性,蛋白結構部分或者全部打開,導致有序結構含量降低,無序結構含量增加。

表1 不同酶處理條件下乳狀液中蛋白質的二級結構相對含量Tab.1 Secondary structure content of protein in oil emulsion with different enzymes treatment %
注:表中同一列不同字母表示數值存在顯著差異(P<0.05)。
3.4.3乳狀液的內源熒光光譜分析
由圖7可知,與原始乳狀液熒光強度(377.82±6.86)相比,所有樣品經堿性蛋白酶和磷脂酶酶解處理后,熒光強度都降低。在溶血磷脂酶酶解后乳狀液中蛋白質的熒光強度最低,這可能與溶血磷脂酶的酶解pH值為4.5有關。經Protex 6L和Acalase 2.4L兩種堿性蛋白酶處理后的乳狀液中蛋白質最大吸收波長和熒光強度降低,并且乳狀液熒光峰發生紅移,可能是經蛋白酶酶解后蛋白結構不穩定,易發生基團移位[32],說明經蛋白酶酶解后界面蛋白的高級結構被破壞,肽鍵斷裂,水解產物的親水性顯著增加,原來處于蛋白質分子內部的色氨酸逐漸暴露于水環境中,其氨基酸殘基所處微環境的極性也逐漸增強,使其疏水性基團的數目減少,不能吸附在油滴表面,進而不能形成界面蛋白膜,而且紅移的程度反映蛋白質構象變化的程度[33],這一結論與表面疏水性數據結果的分析一致。經溶血磷脂酶和磷脂酶D兩種磷脂酶處理導致了光譜的改變,關于不同磷脂酶酶解的產物結構及其不穩定機理有待進一步研究。
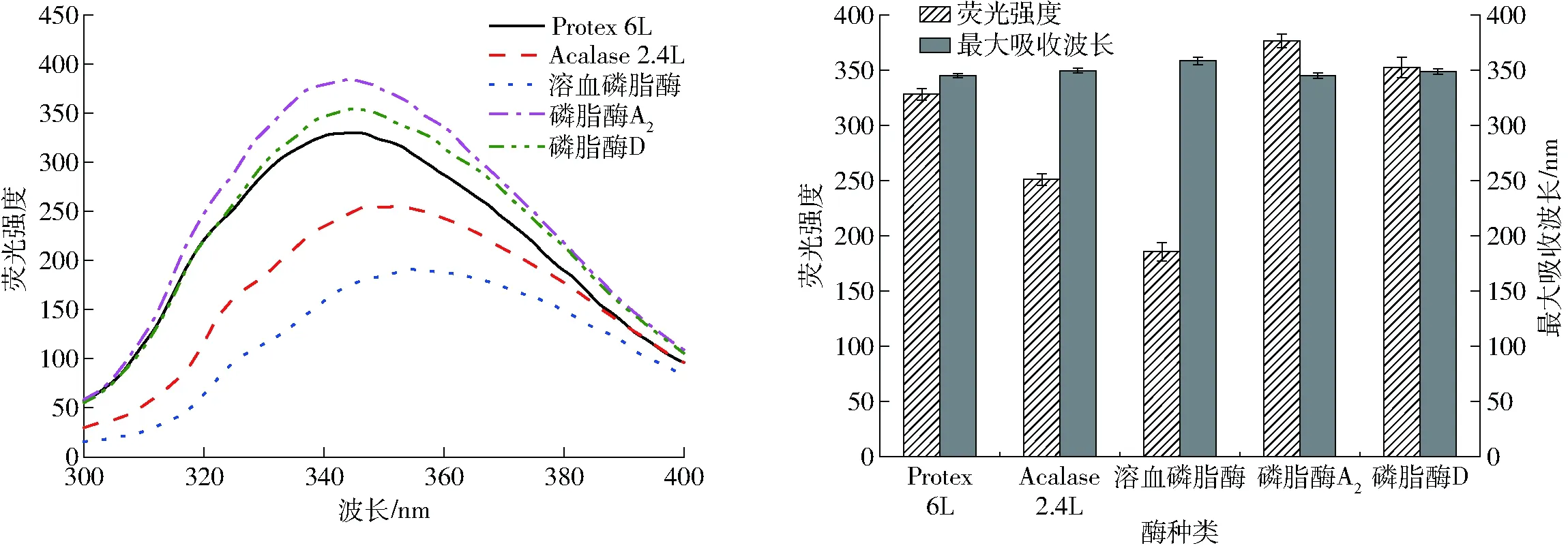
圖7 不同酶處理條件下乳狀液中蛋白質的內源熒光光譜分析Fig.7 Intrinsic fluorescence spectra analysis of protein in oil emulsion with different enzymes treatment
3.5 磷脂酶對乳狀液磷脂的影響
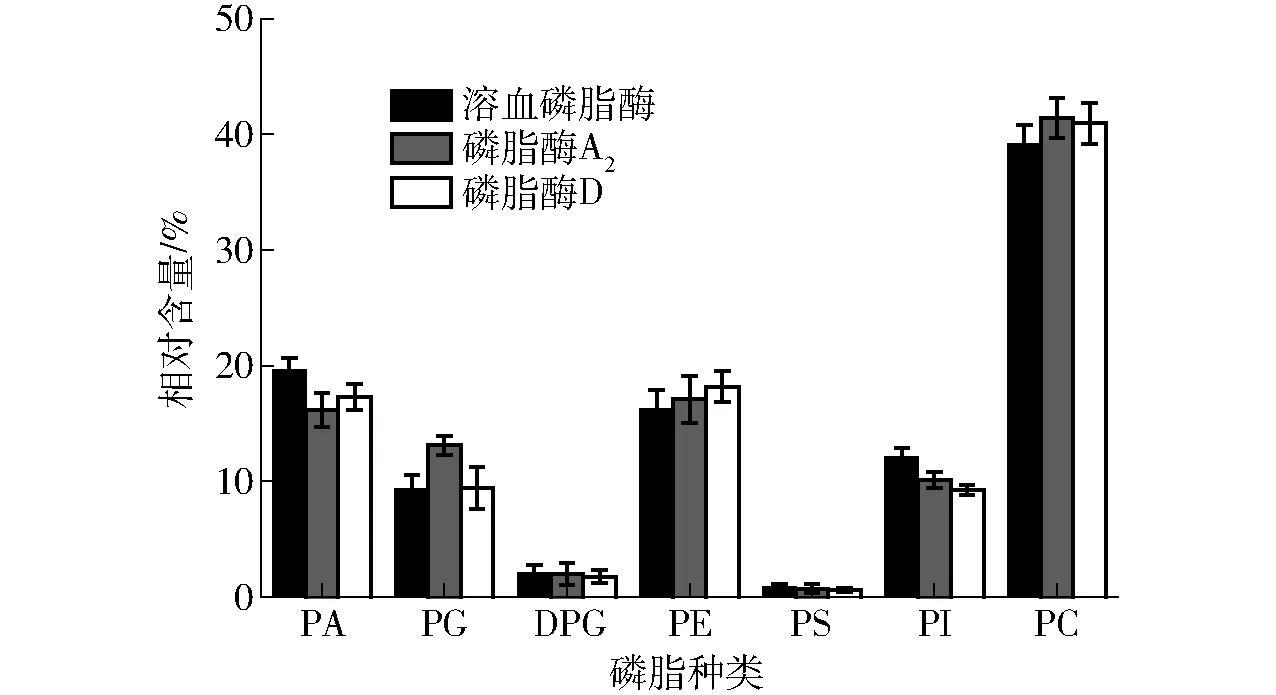
圖8 不同酶酶解乳狀液后磷脂含量31P-NMR圖譜Fig.8 31P-NMR spectrum of phospholipids from oil emulsion with different enzymes treatment
如圖8所示,本研究表明溶血磷脂酶可以切斷酰基磷酸甘油酯的Sn-1鍵產生游離脂肪酸和甘油磷脂酰胺。磷脂酶A2水解磷脂分子中的Sn-2位置得到溶血磷脂和脂肪酸。而磷脂酶D則切斷Sn-4位置的鍵。MENESES等[34]研究發現,磷脂酰膽堿(PC)為大豆粗油中含量最多,且化學位移最不穩定的。原始乳狀液中磷脂主要有磷脂酰膽堿(PC)、磷脂酰乙醇胺(PE)、磷脂酰肌醇(PI)、磷脂酸(PA)、磷脂酰甘油 (PG)、磷脂酰絲氨酸(PS)和雙磷脂酰甘油(DPG),相對含量分別為44.12%、16.4%、11.24%、15.6%、16.4%、0.23%、0.88%。由圖8可知,采用溶血磷脂酶、磷脂酶A2和磷脂酶D酶解乳狀液后,PA含量均升高,PC和PE降低,采用溶血磷脂酶酶解后PA相對含量達到(19.6±1.21)%,比原始乳狀液增長了接近4個百分點,而采用磷脂酶A2和磷脂酶D處理乳狀液后,PA相對含量也分別達到(16.2±1.45)%和(17.3±1.08)%。磷脂酶水解酰基磷酸甘油酯生成磷脂酸和溶血磷脂,使得PA含量升高。采用溶血磷脂酶、磷脂酶A2和磷脂酶D處理乳狀液后,PC相對含量分別降低到(39.12±1.66)%、(41.38±1.73)%和(40.96±1.79)%。PS和DPG含量均升高,但3種酶處理變化不顯著。采用磷脂酶A2處理后的PG含量最高,而溶血磷脂酶處理后的PI含量最高。此外,研究了磷脂酶D,因為它在處理過程中涉及形成不可水解的磷脂,即磷脂酸和溶血磷脂酸,其可影響大豆油質量[35]。
4 結論
(1)采用蛋白酶處理乳狀液時,蛋白酶能水解蛋白生成短肽,分子量變小,表面疏水性降低,蛋白二級結構發生改變,熒光強度降低,進而破壞了乳狀液界面蛋白的完整性,導致在油滴表面的吸附量降低,失去部分電荷,使其不再具有足夠力量阻止油脂的聚集,油滴粒徑增大,解決了生物酶法提取大豆油脂處理時間過長、無法充分破除釋放油脂的問題,油脂提取率得以增加。可以利用這種高效的破乳技術,獲得高提取率的大豆油脂,為生物解離提取大豆油脂產業化提供理論依據。
(2)磷脂酶處理水解了包裹在油脂表面的磷脂,PC和PE水解,PA含量均升高,溶血磷脂、磷脂酶A2酶解Sn-1和Sn-2位后,使磷脂頭部負電性增強,表面所帶的負電荷較多,磷脂酶D的酶解位點為Sn-4位,由于Sn-4位距離磷脂頭部較近,將磷脂頭部呈負電性的磷酸基團酶解成游離的脂肪酸,降低乳狀液的穩定性,油滴的粒徑增大,進而增加了游離油得率。
猜你喜歡
艦船科學技術(2022年16期)2022-09-22 02:15:00
北京航空航天大學學報(2021年6期)2021-07-20 07:23:54
當代陜西(2020年13期)2020-08-24 08:22:02
制造技術與機床(2017年5期)2018-01-19 02:49:17
制造技術與機床(2017年11期)2017-12-18 06:47:29
金秋(2017年4期)2017-06-07 08:22:16
蘇州科技大學學報(自然科學版)(2017年1期)2017-03-20 15:25:18
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
