QuPPe-超高效液相色譜-串聯質譜法測定蔬菜中呋蟲胺及其代謝物殘留
2018-10-08 02:50:30吳延燦蔣冰心施艷紅戚傳勇操海群商魯寧蘇漪玲
食品科學 2018年18期
吳延燦,蔣冰心,施艷紅*,戚傳勇,操海群,唐 俊,商魯寧,蘇漪玲
(1.安徽農業大學植物保護學院,安徽 合肥 230036;2.合肥市農產品質量檢測檢驗中心,安徽 合肥 230091;3.安徽農業大學資源與環境學院,安徽 合肥 230036)
呋蟲胺是日本三井化學公司研發的第三代煙堿類殺蟲劑,與現有的煙堿類殺蟲劑結構相比,它的四氫呋喃基取代了以前的氯代吡啶基、氯代噻唑基,并不含鹵族元素[1-2]。呋蟲胺殺蟲廣譜,主要作用于昆蟲的乙酰膽堿受體,阻斷正常的神經傳遞,從而使昆蟲死亡[3-4],自2002年上市后便廣泛用于防治水稻、蔬菜和水果上的各種害蟲,如飛虱、蚜蟲、葉蟬等[5]。截止2017年7月底,我國批準登記的呋蟲胺產品已達78 個,主要登記使用在水稻、茶樹、番茄、黃瓜、甘藍和馬鈴薯等作物上[6]。研究發現,呋蟲胺會在植物中代謝產生1-甲基-3-[(3-四氫呋喃)甲基]二氫胍鹽(1-methyl-3-(tetrahydro-3-furylmethyl) guanidium dihydrogen,DN)、1-甲基-3-[(3-四氫呋喃)甲基]脲(1-methyl-3-(tetrahydro-3-furylmethyl) urea,UF)等物質,化學結構式見圖1。這些代謝物的毒性與母體大致相同,并且比母體表現出更強的遷移性和持久性[5]。2017年6月18日起實施的GB 2763—2016《食品中農藥最大殘留限量》中[7],僅對呋蟲胺在稻米(2 mg/kg)、糙米(1 mg/kg)、棉籽(1 mg/kg)、黃瓜(2 mg/kg)中的最大殘留限量(maximum residue limits,MRL)值進行了限定,但是對其他登記使用的農作物品種尚未制定MRL值,并且在殘留定義這一項上只明確了呋蟲胺母體,而對代謝物DN、UF并沒有做要求。
目前,國內外報道呋蟲胺的檢測方法主要有酶聯免疫[5]、高效液相色譜法[8-12]和高效液相色譜-串聯質譜法[13-21],但是,同時分析呋蟲胺及其代謝物DN、UF的方法報道較少,目前僅報道了水稻[22-23]、蜂產品[24]、甜瓜[25]以及茶葉[26]上的殘留分析方法,還鮮見蔬菜中呋蟲胺及其代謝物的殘留分析方法報道。近年來呋蟲胺農藥產品在蔬菜上的登記使用越來越廣泛,為有效監測蔬菜中呋蟲胺及其代謝物的殘留情況,保障群眾的消費安全,急需建立蔬菜中呋蟲胺及其代謝物的快速檢測方法。
QuPPe方法是歐盟農藥殘留參考實驗室Anastassiades等[27]于2013年公布的一種采用甲醇提取植物源食品中極性農藥的快速分析方法,目前該方法已經被應用到植物中三唑類殺菌劑[28]、草銨膦[29]和高氯酸鹽[30]等極性化合物的研究中。呋蟲胺及其代謝物屬于極性較大的一類化合物。Rahmana等[26]研究發現甲醇是提取茶葉樣品中呋蟲胺及其代謝物最好的溶劑。因此,本研究采用QuPPe分析超高效液相色譜-串聯質譜(ultra performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)檢測,建立黃瓜、番茄、甘藍、馬鈴薯等登記產品中呋蟲胺及其代謝物DN、UF的殘留分析方法,該方法簡單、高效,能快速、準確地對蔬菜樣品進行定量分析,以期為農產品中呋蟲胺及其代謝物的日常監測和風險評估提供技術保障。
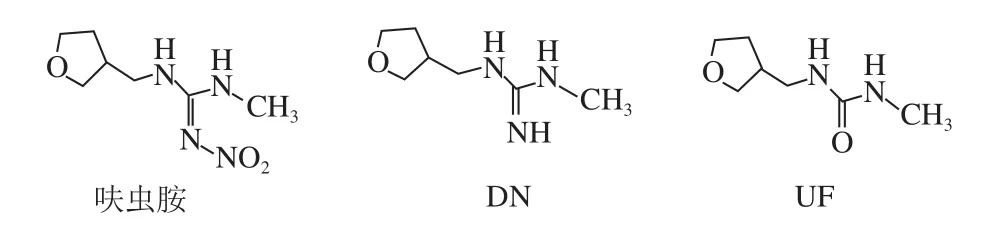
圖1 呋蟲胺及其代謝物DN、UF化學結構式Fig. 1 Chemical structures of dinotefuran, DN and UF
1 材料與方法
1.1 材料與試劑
蔬菜樣品購于合肥市農貿市場。
呋蟲胺(純度99.50%) 德國Dr. Ehrenstorfer公司;DN(純度97.0%)、UF(純度97.0%) 北京勤誠亦信科技開發公司;甲醇、甲酸(均為色譜純) 美國Tedia公司;乙酸(分析純) 國藥集團化學試劑有限公司;實驗用水為密理博超純水系統制備。
1.2 儀器與設備
ACQUITY UPLC Xevo TQ超高效液相色譜-三重四極桿串聯質譜儀(配有電噴霧離子源)、ACQUITY BEH C18色譜柱(2.1 mm×100 mm,1.7 μm) 美國Waters公司;ROTINA380離心機 德國Hettich Zentrifugen公司;VB424多位脈沖渦旋振蕩器 德國Wiggens公司。
1.3 方法
1.3.1 標準溶液配制
分別準確稱取呋蟲胺、DN、UF標準品于10 mL容量瓶中,用甲醇溶解定容配制成質量濃度為1 000 mg/L的母液。同時,用甲醇稀釋成混合標準溶液(100 mg/L),4 ℃條件下保存備用。分別用不同基質的空白提取液逐級稀釋混合標準溶液,配制成1.25、5、12.5、50、125、500 μg/L系列質量濃度的標準工作液。
1.3.2 樣品前處理
準確稱取10.0 g樣品(甘藍取5.0 g)于50 mL離心管中,加入10 mL 1%乙酸的甲醇溶液后渦旋提取2 min(2 500 r/min),以4 000 r/min離心5 min,取上清液到25 mL容量瓶中,用超純水定容至刻度,混勻后過0.22 μm親水性濾膜,供UPLC-MS/MS測定。
1.3.3 UPLC條件
分離采用ACQUITY BEH C18色譜柱(2.1 mm×100 mm,1.7 μm)。流動相A為0.05%甲酸溶液,流動相B為0.05%甲酸-甲醇溶液;梯度洗脫程序:0~0.25 min,70% A;3~4 min,5% A;4.01~5 min,70% A;運行時間5 min;流速0.25 mL/min;柱溫40 ℃;進樣量5 μL。
1.3.4 MS條件
電噴霧離子源,正離子模式;毛細管電壓3.0 kV;離子源溫度150 ℃;錐孔反吹氣流量50 L/h;脫溶劑氣溫度350 ℃;脫溶劑氣流量700 L/h;多反應監測模式。
2 結果與分析
2.1 MS條件的優化
配制1 mg/L呋蟲胺、DN、UF單一標準溶液,在全掃確定母離子后,采用儀器自動調諧模塊對各目標化合物進行質譜條件優化,從而得到最優測定條件,見表1。
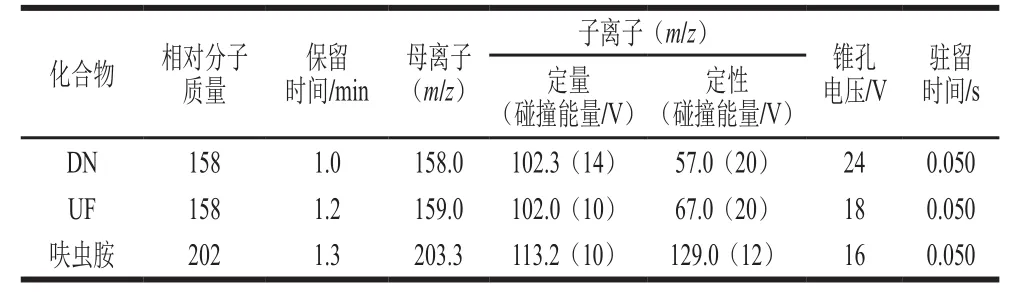
表1 呋蟲胺及其代謝物的測定條件Table 1 Optimized working parameter of ESI-MS/MS for dinotefuran and its metabolites
2.2 色譜條件的選擇
針對極性化合物弱保留的特點,Anastassiades等[27]推薦使用的為常規實驗室不常備的陰離子交換色譜柱(Ionpac AS 11、Ionpac AS 11-HC)以及多孔滲水石墨柱(Hypercarb)。研究發現,采用100 mm規格的ACQUITY BEH C18色譜柱,通過降低流速(0.25 mL/min),調整流動相初始比例(與前處理方法相適應),可以獲得呋蟲胺及其代謝物較好的保留和峰形;加入甲酸可以提高呋蟲胺及其代謝物的離子化效率,從而提高響應,標準溶液色譜圖如圖2所示。
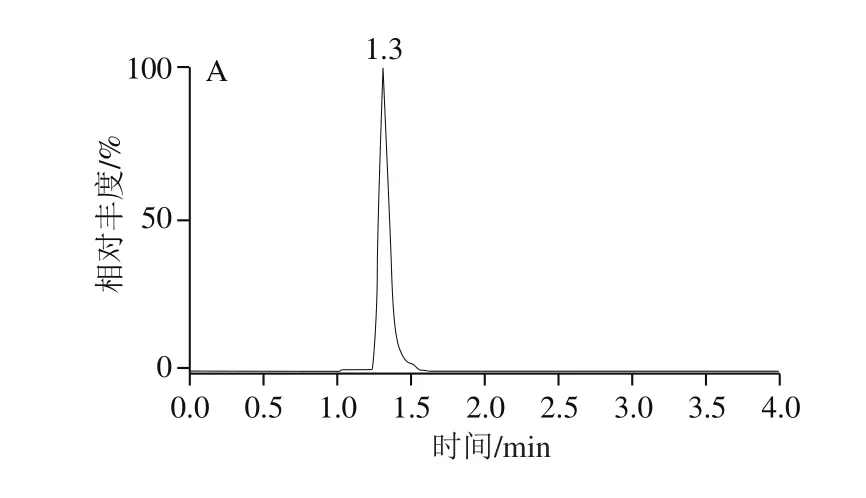
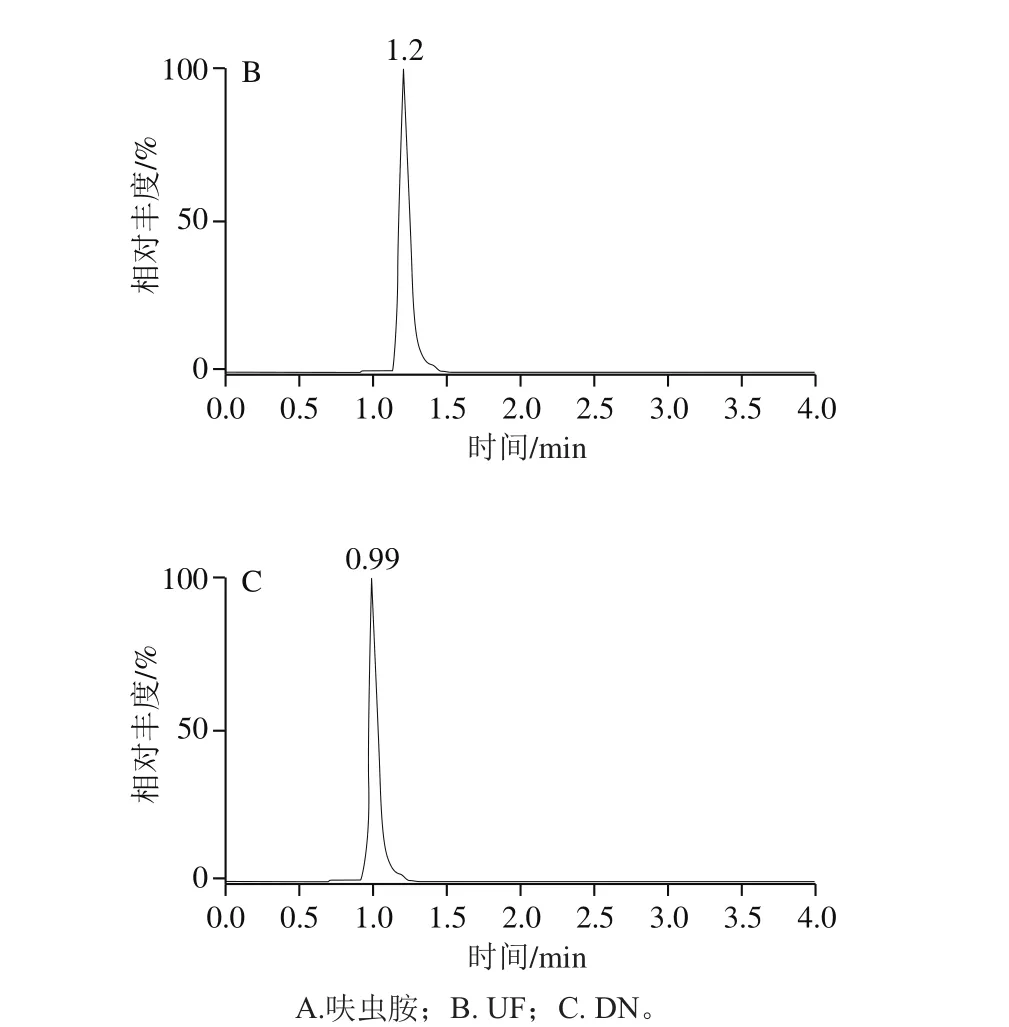
圖2 呋蟲胺及其代謝物定量離子圖譜Fig. 2 Quantitative ion chromatograms of dinotefuran and its metabolites
2.3 提取方法的優化
目前,同時測定呋蟲胺及其代謝物的方法報道較少,大多數方法采用乙腈[26]或乙腈-甲醇混合溶液[22]提取,經過基質固相分散萃取[26]或固相萃取[22]等方式凈化后再測定,步驟較為繁瑣。相對于乙腈,甲醇的共提物更少,提取液幾乎清澈透明,并且對于極性化合物呋蟲胺及其代謝物有更好的提取效率[26],因此本研究借鑒QuPPe方法[27],選用酸化甲醇作為提取劑,采用抗干擾能力強的UPLC-MS/MS聯用測定。蔬菜樣品經提取后,用超純水定容25 mL,不僅保證了待測液與流動相初始比例相似從而獲得更好的峰形,而且稀釋提取液也同時降低基質本底的干擾。甘藍樣品相對于其他樣品提取效率較低,當取樣量和提取劑比例為1∶1時,呋蟲胺和DN的回收率均低于70%,因此本研究將甘藍樣品的取樣量和提取劑比例調整為1∶2。
2.4 線性范圍與檢出限測定結果
用黃瓜、番茄、馬鈴薯、甘藍4 種蔬菜的空白基質提取液配制混合標準工作液(1.3.1節),采用UPLC-MS/MS進行分析,分別以峰面積(y)為縱坐標,質量濃度(x,μg/L)為橫坐標繪制標準工作曲線;對低質量濃度(1.25 μg/L)基質匹配標準溶液進行分析,以3 倍信噪比確定方法的檢出限,10 倍信噪比確定方法的定量限。如表2所示,在1.25~500 μg/L的質量濃度范圍內,呋蟲胺及其代謝物的相關系數均大于0.991 4,方法檢出限為0.04~0.66 μg/L,方法定量限為0.13~2.20 μg/L,見表2。
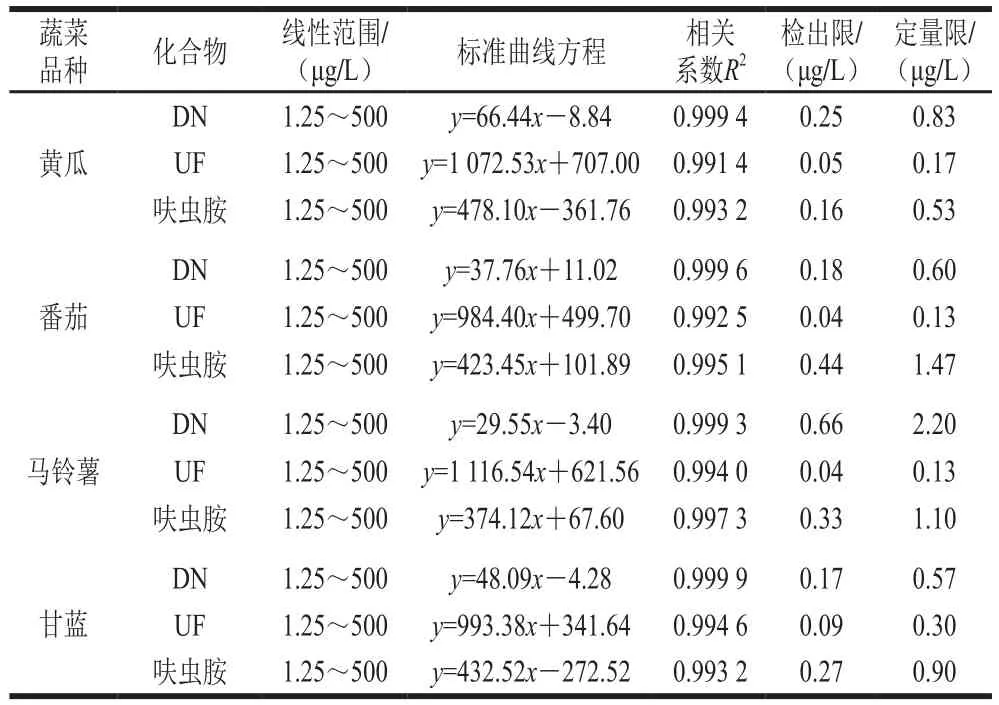
表2 線性范圍與檢出限和定量限Table 2 Linear ranges, linear regression equations, correlation coefficients (R2), LODs, and LOQs
2.5 方法的回收率和精密度結果
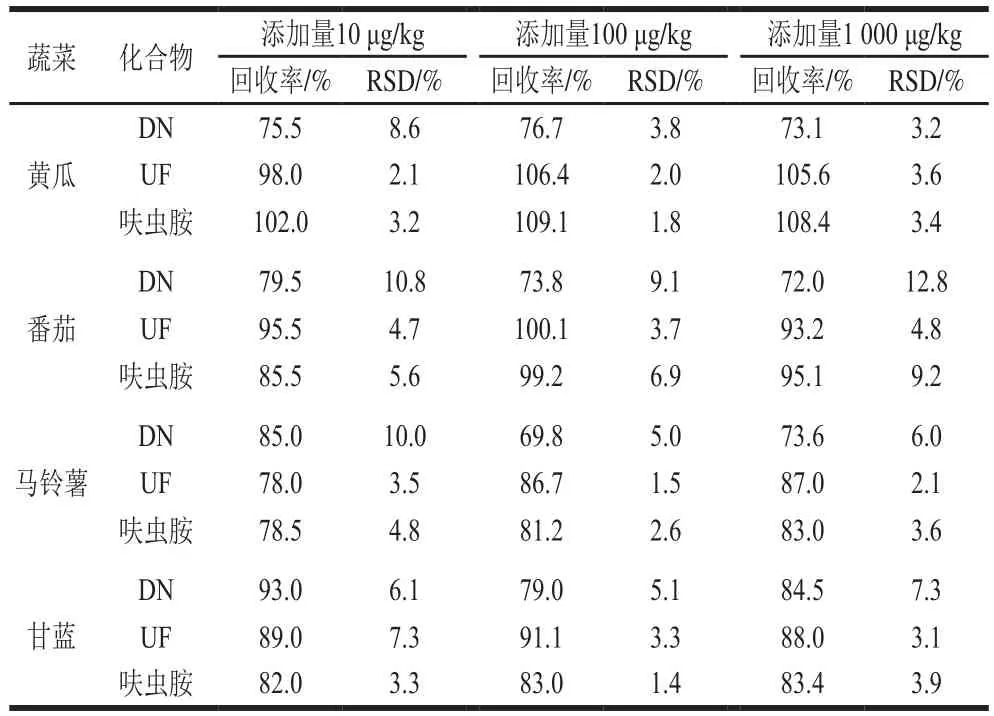
表3 呋蟲胺及其代謝物的加標回收率及精密度(n=5)Table 3 Recoveries and precisions (RSD) of dinotefuran and its metabolites from spiked samples (n= 5)
在空白基質中做添加回收實驗,添加水平分別為10、100 μg/kg和1 000 μg/kg,每個添加水平重復5 次。如表3所示,在黃瓜、番茄、馬鈴薯、甘藍4 種蔬菜中,呋蟲胺及其代謝物的添加回收率為72.0%~109.1%,相對標準偏差(relative standard deviation,RSD)為1.4%~12.8%。呋蟲胺及其代謝物DN、UF在黃瓜、番茄、馬鈴薯、甘藍4 種蔬菜中的最低添加量均為10 μg/kg。
2.6 基質效應測定結果
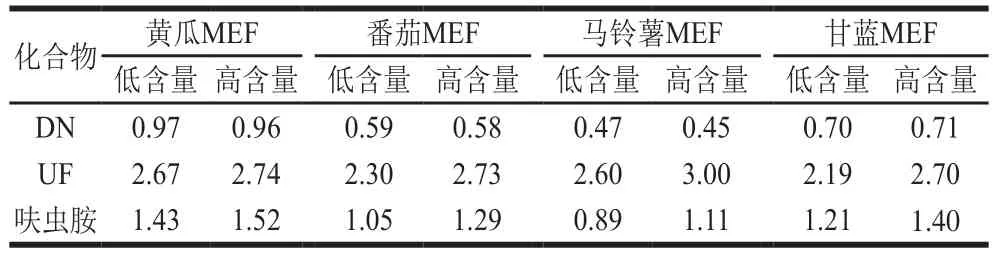
表4 呋蟲胺及其代謝物的基質效應(n=3)Table 4 Matrix effects of dinotefuran and its metabolites (n= 3)
本研究考察黃瓜、番茄、馬鈴薯和甘藍4 種蔬菜基質中呋蟲胺及其代謝物DN、UF的基質效應,分別用溶劑(甲醇-水,2∶3,V/V)和空白基質提取液配制標準溶液,設置12.5 μg/kg(低含量)和125 μg/kg(高含量)以考察不同含量條件下基質效應是否一致,各連續測定3 次。如表4所示,DN在黃瓜基質中表現為弱基質效應(0.8≤MEF≤1.2),在番茄和甘藍基質中表現為中等強度基質效應(0.5<MEF<0.8),在馬鈴薯基質中表現為強基質效應(MEF≤0.5)。UF在4 種蔬菜基質中均表現為強基質效應(1.5≤MEF)。呋蟲胺在馬鈴薯基質中表現為弱基質效應(0.8≤MEF≤1.2);在番茄基質中,低濃度表現為弱基質效應(0.8≤MEF≤1.2),高濃度表現為中等強度基質效應(1.2<MEF<1.5);在甘藍基質中表現為中等強度基質效應(1.2<MEF<1.5);在黃瓜基質中,低濃度表現為中等強度基質效應(1.2<MEF<1.5),高濃度表現為強基質效應(1.5≤MEF)。DN普遍表現為基質減弱效應(MEF≤1),UF和呋蟲胺普遍表現為基質增強效應(1≤MEF)。在本研究設置的2 個含量條件下,UF和呋蟲胺在高含量條件下的基質效應要高于低含量,但是差異不顯著(P<0.05)。考慮到不同蔬菜基質對呋蟲胺及其代謝物結果影響較大,為了保證檢測結果的準確度,在實際樣品的檢測過程中,需要使用基質匹配標準溶液才能有效的補償基質效應對檢測結果的干擾。
2.7 實際樣品的測定
采用本方法對農貿市場所銷售的24 件蔬菜樣品(番茄7 件、黃瓜7 件、馬鈴薯5 件、甘藍5件)中呋蟲胺及其代謝物的殘留量進行分析,其中有1 件黃瓜樣品中檢出呋蟲胺,含量為10 μg/kg,DN含量為117 μg/kg,小于GB/T 2763—2016規定最大殘留限量值2 mg/kg,其他樣品中呋蟲胺及其代謝物未檢出。
3 結 論
目前鮮見針對蔬菜樣品中呋蟲胺及其代謝物檢測方法的報道,而從近期的農藥品種登記情況來看,呋蟲胺產品已經被登記使用在番茄、黃瓜、甘藍、馬鈴薯等蔬菜品種上。本實驗參考歐盟農藥殘留參考實驗室于2013年發布的針對極性農藥的QuPPe分析方法,建立了QuPPe-UPLC-MS/MS同時測定蔬菜中呋蟲胺及其代謝物農藥殘留的方法,該方法簡單、快速,可滿足蔬菜中呋蟲胺及其代謝物的快速篩查和定量分析要求。與傳統方法相比,QuPPe方法更加簡單快速,結合UPLC-MS/MS檢測,可以廣泛應用于一些水果蔬菜基質中極性農藥的快速批量分析。
猜你喜歡
核科學與工程(2021年4期)2022-01-12 06:30:26
今日農業(2020年19期)2020-12-14 14:16:52
小學生必讀(中年級版)(2020年9期)2020-12-04 02:07:22
兒童故事畫報(2019年5期)2019-05-26 14:26:14
中學物理·高中(2016年12期)2017-04-22 11:53:03
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
小櫻桃·童年閱讀(2014年11期)2014-12-01 22:21:30
