JAK2突變與骨髓增生異常綜合征的關系
2018-10-17 09:42:32周旭艷牧啟田歐陽桂芳
浙江醫學 2018年19期
關鍵詞:信號
周旭艷 牧啟田 歐陽桂芳
Janus激酶(JAKs)是一類非受體型可溶性的胞質蛋白酪氨酸激酶,共有4個成員:JAK1、JAK2、JAK3和TyK2,大約介導60種細胞因子和激素信號,細胞外信號與激酶的相互作用決定了JAKs的生物學功能[1]。JAK2是JAKs中與血液疾病關系最為密切的激酶,JAK2基因突變可影響骨髓造血細胞的信號傳導通路,對血液疾病的發生、進展意義重大[2]。目前JAK2-V617F突變被認識到與骨髓增殖性疾病(MPDs)關系最密切[3],然而多項研究發現JAK2突變在骨髓增生異常綜合征(MDS)的惡性發病機制中也起到重要作用,伴有JAK2突變的MDS患者往往預后不良[4-8]。本文就JAK2、JAK2/STAT信號通路及JAK2與MDS的關系等綜述如下。
1 JAKs的結構和活性改變
JAKs由7個不同長度的JAK同源區(JAK homology,JH)(JH1-JH7)構成,包含 4 個功能區域,分別是 N-端的FERM區、SH2區、假酪氨酸激酶結構域(pseudokinase,PK)和C-端催化活性氨基酸激酶結構域(tyrosine kinase,TK)。FERM區由JH5-JH7和小部分JH4組成,調節催化活性,此區域的突變會改變JAKs蛋白激酶活性。SH2區包括大部分JH4和小部分JH3,對JAKs的活化起輔助作用。FERM區和SH2區共同作用激酶與其同源受體的相互結合,影響胞內信號傳導。非功能性PK區(JH2)負性調節JAKs蛋白激酶活性,JH2功能的缺失影響細胞因子誘導的通路信號,造成機體嚴重的聯合免疫缺陷。TK區位于JH1,具有催化活性,此區域位點的突變并不多見。最常見的JAK2-V617F突變發生于JH2同源區,JAK2-V617F突變時JH2結構區構象隨之發生改變,對JH1的抑制作用減弱,使JH1磷酸化而被持續激活,從而異常啟動下游信號通路[1,9],見圖1。
JAKs的活性受到JAKs自身磷酸化位點和與之作用的蛋白調節。自身磷酸化位點包括負性調控位點和正性調控位點,其中負性調控位點包括Ser523、Tyr317、Tyr570 等,正性調控位點包括 Tyr1007、Tyr1008、Tyr221、Tyr813等。影響JAKs活性的蛋白包括SHP1、SHP2、PTP1B、TCPTP、CD45等蛋白質酪氨酸磷酸酶,它們通過使JAKs位點發生脫磷酸化,而影響后者活性。此外SOCS1蛋白、SOCS3蛋白和SH2B家族蛋白質對JAKs有直接的負性調控作用[1],見圖1。
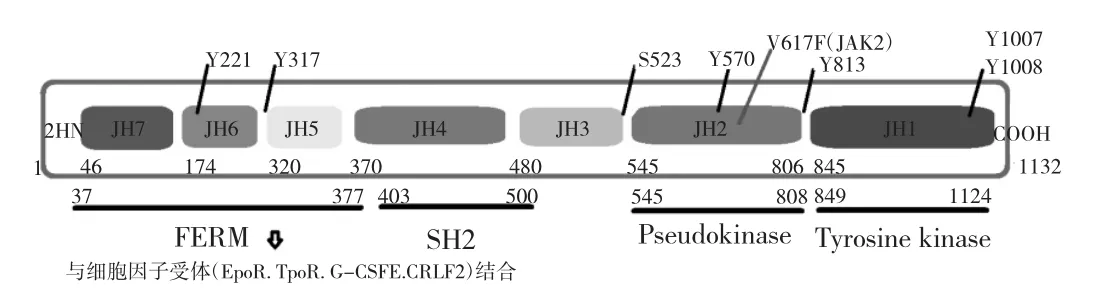
圖1 JAKs結構模式圖
2 JAK2基因突變類型
JAK2基因長 140kb,位于染色體 9p24,編碼由1 132個氨基酸組成的信號轉導蛋白,JAK2的突變會影響骨髓造血細胞的信號傳導通路。JAK2基因點突變中以JAK2-V617F(JAK2基因第14號外顯子編碼序列的第1 849位堿基G被T取代,導致JH2假激酶區617位置纈氨酸轉變為苯丙氨酸)突變率最高,多見于真性紅細胞增多癥(polycythemia vera,PV)[3]。JAK2-V617F等位基因負荷超過50%是PV患者病情進展、合并骨髓纖維化(myelofibrosis,MF)的高危因素[10]。而 MDS 患者的JAK2突變也以JAK2-V617F最為常見。其他類型點突變如JAK2-R683S(G)多見于急性B淋巴細胞白血病[11-12],總體發生率低。JAK2的12號外顯子堿基的插入和缺失突變多見于PV,一般發生在第537至543位密碼子之間,其中以N542-E543del的發生率最高(30%)。相比于JAK2-V617F突變,12號外顯子異常的患者血象中Hb水平更高、PLT和WBC數更低,但血栓形成、繼發骨髓纖維化和病情進展的概率相似[13]。JAK2基因重排的類型包括TEL/ETV6-JAK2、PCM1-JAK2、BCR-JAK2等,在MDS患者中的發生頻率低。JAK2基因擴增在血液惡性腫瘤中非常少見,在MDS患者中未見報道。
3 JAK2與信號傳導及轉錄激活因子途徑(STAT)
人類STAT家族共有6個成員:STAT1、STAT2、STAT3、STAT4、STAT5(STAT5a、STAT5b)、STAT6,以 STAT3和STAT5蛋白與淋巴和骨髓惡性腫瘤的發病機制最為相關。STAT5包含STAT5a和STAT5b兩種蛋白,主要在造血干、祖細胞和成熟細胞群的信號轉導中發揮作用[14]。破壞STAT3和STAT5之間的生理平衡也可導致血液惡性腫瘤的發生[15]。
JAK/STAT途徑本質上是一個超過30種跨膜蛋白的超家族。作為細胞因子受體信號轉導的中心,JAK2/STAT可識別特定的細胞外信號因子,向胞內傳遞增殖、分化、抗凋亡等信號,對造血系統、免疫反應的調節至關重要[16]。JAK2的突變狀態可影響信號傳導。當JAK2為野生型時,只有與細胞外配體結合,才能激活細胞內STAT、PI3K和RAS-MAPK等信號通路,進而調節基因轉錄和細胞的增殖、分化;當JAK2發生突變時,即使沒有細胞外任何配體,也可持續性激活STAT等多條信號通路[16],見圖2。除JAK2自身發生的突變外,負性調節因子的突變或沉默、JAK2致癌融合蛋白的存在及調控因子含量的改變等均可激活STAT途徑。國外一項研究用小鼠模型觀測了JAK/STAT信號通路與負性調控因子CD45和靶分子轉移蛋白受體CD71之間的關系,結果發現低表達的CD45和CD71致JAK/STAT通路持續性激活,最終導致MDS造血異常的發生[17]。

圖2 野生型JAK2和突變型JAK2信號通路激活模式圖
4 JAK2突變與MDS的關系
4.1 JAK2的突變率 一般認為JAK2基因以JAK2-V617F突變的形式發生于MPDs最為常見,其中在PV患者中的突變率約為95%,原發性血小板增多癥和原發性MF患者中的突變率分別約為50%,而在MDS和急性髓細胞白血病患者中的突變率<5%[18-19]。
陳婉等[4]報道在中國人群中,MDS總體的突變陽性率為5%,難治性血細胞減少伴多系發育異常、難治性貧血伴原始細胞增多和不能分類的MDS亞型均檢出JAK2-V617F突變,各亞型之間頻率并無差異。Jekarl等[20]在檢測非骨髓增殖性腫瘤的血液疾病患者基因突變時發現,MDS中JAK2-V617F突變率為8.3%。這些結果提示,盡管在MDS中JAK2突變率并不是很高,但與MDS的發病機制有一定相關性。
4.2 JAK2突變與MDS病情進展及預后 JAK2突變在MDS病情進展中起到一定作用。伴JAK2-V617F突變的MDS患者骨髓原始細胞比例、外周血乳酸脫氫酶水平高,脾臟腫大發生率高,并且約有37.5%的JAK2突變患者會發生急性白血病轉化,遠高于野生型患者(5.9%)[4]。此外,JAK2突變可以在MDS轉白期間獲得,并且可能在低危MDS亞型的病情進展中起作用[5]。國內一項臨床研究對比了MDS患者和健康志愿者外周血JAK2-V617F基因的表達水平,結果發現JAK2-V617F基因表達拷貝數在惡性程度高的MDS類型中明顯升高,提示MDS的發病可能受JAK2信號通路異常的影響[6]。綜合上述結果,JAK2-V617F突變水平檢測可作為MDS病情評價的一個指標。
JAK2突變與MDS合并MF也有密切關系。研究發現,在MDS合并MF患者中JAK2-V617F突變的發生率(21%)遠大于無MF者(2%)。臨床中合并MF的患者更易發生肝脾腫大,更傾向于出現消耗性癥狀和輸血依賴,其生存率明顯偏低,并且病情進展速度快[7]。Fu等[7]發現MDS合并骨髓纖維化患者JAK2-V617F等位基因負荷的中位值為22.5%(5.5%~46%),低于原發性骨髓纖維化(47%)。因此,檢測JAK2-V617F的基因負荷有助于區別原發和繼發性骨髓纖維化,基因負荷高的骨髓纖維化患者更傾向于原發性骨髓纖維化,若在MDS患者中檢測到高負荷的JAK2-V617F突變需警惕合并骨髓纖維化可能,提示預后不良。盡管MDS合并骨髓纖維化的JAK2-V617F等位基因負荷明顯低于原發性骨髓纖維化,但JAK2-V617F突變的存在有可能加速MDS患者骨髓纖維化的進程,同時影響骨髓移植的治療效果。臨床資料顯示JAK2突變也是MDS患者移植后存活率低的預測因子之一,當MDS患者伴有JAK2突變時,移植后生存期明顯低于無JAK2突變者,在需要骨髓移植的MDS患者中檢測JAK2突變對評估預后有益[8]。
4.3 JAK2突變與MDS的治療 目前MDS治療方案主要包括支持治療、低甲基化劑(阿扎胞苷、地西他濱等)、生物反應調節劑和免疫抑制劑(抗胸腺細胞球蛋白、環孢素和來那度胺等)、化療及異基因造血干細胞移植等[21-24],但上述治療方案在臨床上仍存在諸多問題,因此,針對JAK2及其信號通路的靶向治療是MDS潛在治療手段之一。
盧索替尼是一種可口服的選擇性JAK1/JAK2激酶抑制劑,是首個被美國FDA批準用于治療中高危MF患者的藥物。在多組臨床研究中證實盧索替尼可明顯縮小原發性MF患者的脾臟體積,改善患者的臨床癥狀和生活質量[25-26]。在PV患者中,盧索替尼有明顯控制血細胞比容、減小脾臟體積、減少血栓栓塞事件發生的作用[27]。此外,有相關報道稱,伴有JAK2基因融合的骨髓增殖性腫瘤、慢性白血病以及骨髓增生異常綜合征/骨髓增殖性腫瘤的患者在使用盧索替尼后,腫瘤負荷獲得明顯的改善[28-29]。但目前臨床上對盧索替尼治療伴有JAK2基因突變或合并MP的MDS患者的療效尚不明確。Junya等[30]發現JAK2選擇性抑制劑NS-018可優先抑制MDS-BMMNC(骨髓單核細胞)的增殖,有效預防MDS患者惡性克隆的過度增殖,并且對正常BMMNC產生的毒性較低。因此,NS-018有望被運用于臨床治療具有增殖性表型高危的MDS患者。
Boehrer等[31]通過體外細胞實驗發現表皮生長因子受體抑制劑厄洛替尼具有抗腫瘤活性。厄洛替尼能夠通過脫靶抑制作用破壞JAK2/STAT5通路的信號傳導,殺死來自MDS和急性髓細胞白血病患者的CD34+骨髓原始細胞,同時能夠保留正常的CD34+祖細胞。
研究發現,通過單藥地西他濱化療可降低MDS患者JAK2-V617F突變的表達水平,并且通過增加化療周期可使拷貝數進一步降低[32]。JAK2突變的高表達可能導致MDS發生從低型向高危型惡性轉化,去甲基化藥物對此機制有抑制作用,而JAK2-V617F拷貝數的變化或許可以預示去甲基化藥物的治療效果。
西達本胺是一種新型苯甲酰胺類的組蛋白去乙酰化酶抑制劑,具有抗腫瘤活性[33]。Zhao等[34]在細胞實驗中證實了這種抗腫瘤活性也作用于MDS,西達本胺可上調SOCS3的表達水平從而抑制MDS細胞JAK2/STAT3信號通路的傳導,并且可能通過JAK2/STAT3傳導信號的下調誘導MDS細胞G0/G1期停滯及凋亡。因此,西達本胺除了用于治療難治復發的外周T細胞淋巴瘤外,還有治療MDS的可能性,這一觀點也得到其他研究者的支持[35]。
5 小結與展望
JAK2突變與MPDs的關系已經廣為臨床認可。JAK2-V617F突變在MDS的惡性發病機制中起到重要作用,且此類MDS患者具有獨特的臨床表現,往往預后不良。JAK2突變及其信號通路異常與MDS的關系是目前臨床關注的熱點。
安全有效的JAK2及其信號通路抑制劑正在不斷地被研發,并取得了較好的臨床效果。盡管如此,JAK2抑制劑治療伴JAK2基因突變MDS患者的分子機制和臨床有效性研究仍然少見。目前已發現多種治療MDS藥物的起效機制可能與JAK2/STAT途徑的改變有關,但此類研究多在細胞實驗中進行,臨床試驗少見。因此,真正實現把JAK2及其信號通路作為靶點來治療MDS這一目標,仍任重而道遠。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
