氯離子通道蛋白1的缺失突變體CLIC1(Δ49-51)與Sedlin蛋白相互作用的研究
2018-11-07 05:38:04洪翠麗金振輝朱亮亮潘林鑫耿慧武劉曉穎
安徽醫科大學學報 2018年11期
洪翠麗,金振輝,朱亮亮,潘林鑫,耿慧武,劉曉穎
氯離子通道蛋白1(chloride intracellular channel 1,CLIC1)是氯離子通道家族中的一員。CLIC家族中對CLIC1的研究較多,其可以插入膜并形成氯離子通道。CLIC1的膜結合形式中N末端是細胞膜結合位點,C末端在膜內側[1-2]。CLIC1的C末端與谷胱甘肽S轉移酶(glutathione-S-transferase, GST)具有很高的同源性[3-4];N末端是高度保守區域,含有硫氧還原蛋白區域,可折疊成谷氧還原蛋白,其中CLIC1中的Trp35(色氨酸殘基)被認為是CLIC1蛋白的跨膜結合位點[5-6]。以往的研究[7-8]表明CLIC1與神經性疾病、惡性腫瘤增殖相關。 據報道CLIC1的第49~51位3個氨基酸是跨膜結構域中最重要的序列[9]。在本課題組前期已經證實了CLIC1與Sedlin(是SEDL基因的表達產物)在哺乳動物細胞內存在相互作用[10]的基礎上,構建第49~51位氨基酸的缺失突變體重組質粒pcDNA3.1-CLIC1(Δ49-51)-FLAG,進一步研究CLIC1(Δ49-51)與Sedlin在HEK 293T細胞中是否存在相互作用,探究這3個氨基酸的缺失對其與Sedlin相互作用的影響,為進一步揭示CLIC1在腫瘤中的作用機制奠定基礎。
1 材料與方法
1.1材料
1.1.1細胞、質粒和菌株 載體pcDNA3.1、pGEX-5X-3,菌株TG1、BL21、DH5α等,重組質粒pcDNA3.1-CLIC1-FLAG和pcDNA3.1-CLIC1(Δ49-51)-FLAG均為本實驗室保存。COS7細胞、HEK 293T細胞等也為本實驗室常用細胞。
1.1.2主要試劑 PrimeSTAR 酶購自日本TaKaRa公司;BamH I、Xho I、EcoR I等限制性內切酶;質粒提取試劑盒購自美國OMEGA公司;蛋白Marker購自美國Thermo公司;胎牛血清購自美國Hyclone公司;LipofectamineTM2 000、Opti-MEM購自美國Invitrogen公司;FLAG抗體購自美國Sigma公司;熒光封片膠購自丹麥DAKO公司;PVDF膜購自加拿大BioBasic公司;SuperSignal West Pico 顯色試劑盒購自美國Pierce公司;激光共聚焦顯微鏡由安徽醫科大學大型儀器共享平臺提供;引物由生工生物工程(上海)股份有限公司合成。
1.2方法
1.2.1細胞培養和質粒轉染 用DMEM高糖培養基(其中含5%的胎牛血清和100 U/ml的青霉素和鏈霉素)培養COS7細胞和HEK 293T細胞,其中培養箱為37 ℃、5% CO2;當細胞長滿單層時傳代,PBS洗2次,胰酶消化至細胞變圓時,DMEM終止消化;離心后用PBS緩沖液重懸細胞清洗,并以適當密度接種到培養皿中,過夜培養;次日,將質粒(μg) ∶脂質體(μl)按1 ∶2.5的比例轉染HEK 293T細胞,用于Western blot實驗。而免疫熒光實驗按1 ∶1.5[質粒(μg) ∶脂質體(μl)]的比例轉染,轉染4~6 h換新鮮的培養液(不含雙抗)。
1.2.2免疫熒光爬片 轉染24 h后取出培養皿,用放置在4 ℃冰箱的PBS緩沖液洗3次;-20 ℃預冷的甲醇固定2 min,70%的乙醇固定5 min;1%脫脂奶粉(用TBST配制)封閉30 min;先用FLAG抗體孵育2 h,隨后山羊抗小鼠IgG(TRITC標記的)孵育1 h;DAPI染核3 min后,PBS洗3次,每次5 min;熒光封片膠DAKO封片;次日觀察并拍攝激光共聚焦照片。
1.2.3GST pulldown實驗 同上述轉染方法,轉染48 h后收細胞,棄去培養液,用PBS小心洗1次,棄去PBS,然后在培養皿中加入適量胰酶,胰酶消化1 min左右(顯微鏡下觀察到細胞從壁上脫落下來),低速離心(1 500 r/min 、2 min)收集細胞于1.5 ml EP管中,加入適量細胞裂解液(按細胞量的多少具體情況而定),4 ℃ 層析柜混旋儀上混懸40 min(收細胞的前1 d下午用預溫的PBS清洗GST珠子3次,分別加入GST和GST-Sedlin融合蛋白混旋儀上過夜混懸),把裂解好的細胞裂解液取出40 μl加入10 μl的5×SDS混合,煮沸,冰浴,放于-20 ℃ 冰箱待電泳。把剩下的細胞裂解液分別加入到上述過夜混懸好的GST和GST-Sedlin融合蛋白(過夜混懸好結合緩沖液洗3次)里,再次混懸4 h。混懸結束后(結合緩沖液再次洗3次)加入等量的2×SDS,煮沸7 min,冰上5 min,下一步Western blot分析。
1.2.4免疫共沉淀實驗 同上述轉染方法,轉染48 h后收細胞,棄去培養液,用PBS小心洗1次,棄去PBS,然后在培養皿中加入適量胰酶,胰酶消化1 min左右(顯微鏡下觀察到細胞從壁上脫落下來),低速離心(1 500 r/min、2 min)收集細胞于1.5 ml EP管中,加入適量細胞裂解液(按細胞量的多少具體情況而定),4 ℃ 層析柜混旋儀上混懸40 min,把裂解好的細胞裂解液分別取出40 μl加入10 μl的5×SDS混合,煮沸,冰浴,放于-20 ℃冰箱待電泳。把剩下的細胞裂解液分別加入適量的FLAG抗體再次混懸4 h,混懸結束后把混懸好的混合液加入到事先清洗好的瓊脂糖珠子里(混懸快結束時用預溫的PBS清洗瓊脂糖珠子3次),再次混懸4 h,混懸結束后用IP清洗液再次洗結合珠子3次(4 ℃、3 000 r/min,3 min)棄上清液,加入等量的2×SDS,煮沸7 min,冰上5 min,下一步Westen blot分析。
1.2.5Westen blot分析 配制12%的分離膠和5%的濃縮膠,電泳先30 min 70 V后80 min 100 V。三明治裝置濕轉100 V、75 min。取出PVDF膜,封閉液(TBST+5%脫脂奶粉配制)封閉2 h,FLAG鼠抗(1 ∶500)4 ℃孵育過夜,FLAG二抗(1 ∶5 000)孵育1.5 h,顯影。
1.2.6核質分離實驗 轉染48 h后收細胞,胰酶消化2 min,將細胞收集到離心管中并加入5倍體積的CE buffer,同時冰上裂解6 min,間歇震蕩裂解液。4 ℃、3 000 r/min 離心5 min,收集上清液。加入相等量的2×SDS上樣緩沖液于上清液中,沸水浴7 min,冰上冷卻后,-20 ℃存放備用。用適量的CE buffer(不含NP-40)洗滌沉淀,4 ℃、3 000 r/min 離心5 min,用同樣的方法洗滌2~3次以確保除盡沉淀中的胞質蛋白。加入與之相等體積的NE buffer于剩余的沉淀中,冰上裂解11 min,間歇震蕩裂解液。4 ℃、14 000 r/min 離心6 min,收集上清液,加入適量5×SDS上樣緩沖液,沸水浴7 min,冷卻后,-20 ℃存放備用。
2 結果
2.1CLIC1缺失突變體的構建對質粒pcDNA3.1-CLIC1(Δ49-51)-FLAG進行雙酶切(BamH I, Xho I),見圖1。CLIC1缺失突變體被切成2條強帶,與Marker對比,一條帶大約在5 148 bp位置上,另一條位置不超過831 bp,相應的質粒送往通用生物有限公司進行測序,反饋的結果指示質粒構建成功。

圖1 重組質粒酶切鑒定圖和測序圖
M:DNA Marker;1:pcDNA3.1-CLIC1-FLAG的酶切鑒定結果;2:pcDNA3.1-CLIC1(Δ49-51)-FLAG的酶切鑒定結果和測序圖
2.2CLIC1及其缺失突變體CLIC1(Δ49-51)蛋白在HEK293T細胞中的表達Western blot結果顯示,轉染了表達質粒pcDNA3.1-CLIC1-FLAG和pcDNA3.1-CLIC1(Δ49-51)-FLAG的HEK 293T細胞裂解液分別能檢測到相應的條帶。與預染蛋白Marker相比較其符合CLIC1蛋白和CLIC1(Δ49-51)蛋白的大小。CLIC1蛋白大小為27 ku,CLIC1(Δ49-51)蛋白的分子量略低于27 ku。見圖2。
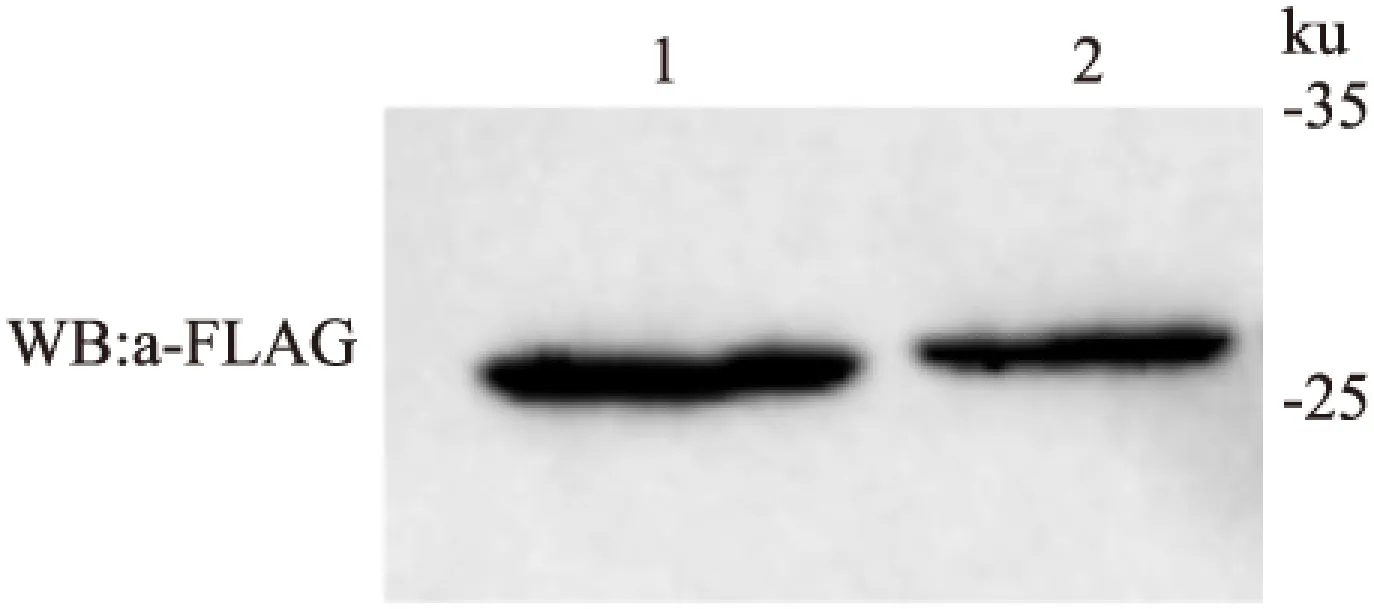
圖2 Western blot檢測 CLIC1(Δ49-51)和CLIC1蛋白在哺乳動物細胞中的表達
1:轉染pcDNA3.1-CLIC1(Δ49-51)-FLAG的細胞裂解液;2:轉染pcDNA3.1-CLIC1-FLAG的細胞裂解液
2.3CLIC1(Δ49-51)蛋白在COS7細胞中的定位以及其與Sedlin蛋白的共定位CLIC1(Δ49-51)蛋白在COS7細胞的細胞核和細胞質中均有明顯的表達,且在細胞質表達量明顯多于細胞核,見圖3A。圖3B示CLIC1和Sedlin的共定位。CLIC1(Δ49-51)蛋白與Sedlin蛋白在COS7細胞的細胞核、細胞質中均不存在共定位現象,見圖3C。
2.4CLIC1(Δ49-51)與Sedlin蛋白在體外不存在相互作用分別對CLIC1、CLIC1(Δ49-51)蛋白與Sedlin蛋白進行了GST pulldown實驗,即對照組[GST+CLIC1-FLAG/GST+CLIC1(Δ49-51)-FLAG]和實驗組[GST-Sedlin+CLIC1-FLAG/GST-Sedlin+CLIC1(Δ49-51)-FLAG],收集谷胱甘肽珠子進行Western blot分析,見圖4A、4B。進行免疫印跡(用FLAG抗體)后,A中1泳道細胞裂解液組和3泳道實驗組均出現了明顯條帶,而B中3泳道實驗組和2泳道對照組均未出現任何條帶。并且1、3泳道的條帶位置基本一致,與預染蛋白Marker 27 ku的條帶橫向比對后,其大小分別與CLIC1(27 ku)和CLIC1(Δ49-51)(27 ku)的大小相符。同時,又對A、B中的1、2、3的樣品進行GST抗體免疫印跡后,見圖4A、4B,對照組(2泳道)和實驗組(3泳道)分別約在Marker的20~30 ku和30~45 ku之間的位置存在很強的蛋白條帶,大小分別與GST(26 ku)和GST-Sedlin(42 ku)融合蛋白的大小相符。綜上表明,CLIC1蛋白與Sedlin蛋白在體外存在相互作用,而CLIC1(Δ49-51)蛋白與Sedlin蛋白在體外不存在相互作用。
2.5CLIC1(Δ49-51)與Sedlin蛋白不存在相互作用在上述GST pulldown體外實驗的基礎上,進行了相應的免疫共沉淀實驗:CLIC1-FLAG+GFP/CLIC1(Δ49-51)-FLAG+GFP(對照組),CLIC1-FLAG+GFP -Sedlin/CLIC1(Δ49-51) -FLAG+GFP-Sedlin(實驗組),見圖5A、5B。進行免疫印跡(用FLAG抗體)后,對照組1、2泳道和實驗組3、4泳道均出現了明顯的條帶,與預染Marker比對,目的條帶在25~35 ku之間,1~4號泳道條帶的大小與CLIC1及CLIC1(Δ49-51)(27 ku)的蛋白大小相符。免疫印跡(用GFP抗體)后,對照組1、2泳道和A中實驗組4泳道均出現了明顯條帶,與預染Marker橫向比對,各目的條帶的分子量大小正確,而對照組3泳道和B中實驗組4泳道未出現任何條帶。該結果顯示,在HEK 293T細胞中,CLIC1蛋白與Sedlin蛋白存在相互作用,而CLIC1(Δ49-51)蛋白與Sedlin蛋白不存在相互作用。
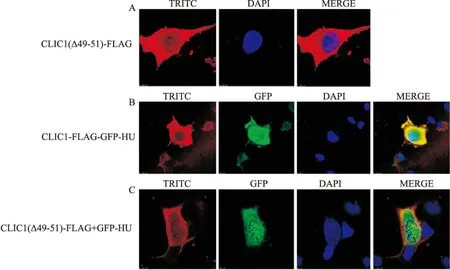
圖3 CLIC1(Δ49-51)在COS7細胞中的定位及其與Sedlin蛋白的共定位
TRITC:FLAG標簽蛋白;DAPI:示細胞核;GFP:GFP標簽蛋白;MERGE:疊加效果;A:CLIC1(Δ49-51)蛋白在COS7細胞中的定位;B: CLIC1與Sedlin蛋白在COS7細胞中的共定位; C: CLIC1(Δ49-51)與Sedlin蛋白在COS7細胞中的共定位
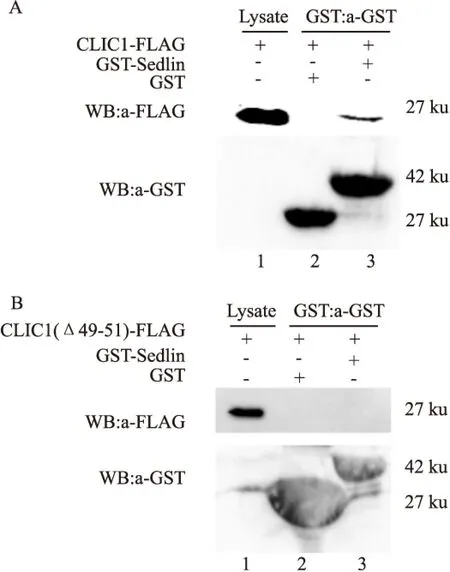
圖4 CLIC1、CLIC1(Δ49-51)與Sedlin蛋白的GST pulldown實驗
1:單轉CLIC1-FLAG/CLIC1(Δ49-51)-FLAG的細胞裂解液; 2:谷胱甘肽珠子下拉GST+CLIC1-FLAG/GST+CLIC1(Δ49-51)-FLAG混合液; 3:谷胱甘肽珠子下拉GST-Sedlin+CLIC1/GST-Sedlin+CLIC1(Δ49-51)-FLAG混合液
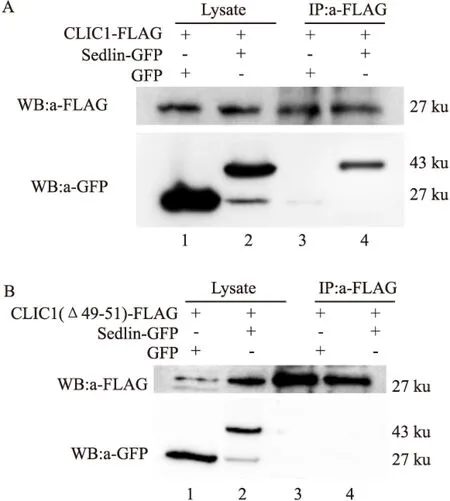
圖5 CLIC1、CLIC1(Δ49-51)與Sedlin蛋白的免疫共沉淀實驗
1:共轉CLIC1-FLAG+GFP/CLIC1(Δ49-51)-FLAG+GFP的細胞裂解液; 2:共轉CLIC1-FLAG+Sedlin-GFP/CLIC1(Δ49-51)-FLAG+Sedlin-GFP的細胞裂解液; 3:用FLAG抗體免疫沉淀共轉CLIC1-FLAG+GFP/CLIC1(Δ49-51)-FLAG+GFP的細胞裂解液; 4:用FLAG抗體免疫沉淀共轉CLIC1-FLAG+GFP-Sedlin/CLIC1(Δ49-51)+Sedlin-GFP的細胞裂解液
2.6CLIC1(Δ49-51)蛋白與CLIC1蛋白在COS7細胞中表達存在差異在上述實驗的基礎上,進行了CLIC1、CLIC1(Δ49-51)的核質分離實驗,見圖6,進行免疫印跡(FLAG抗體)后,1~4泳道均出現了明顯的條帶,與預染Marker比對,目的條帶在25~35ku之間,1~4號泳道條帶的大小與CLIC1及CLIC1(Δ49-51)(27ku)的蛋白大小相符。以Lamin抗體免疫印跡后,2泳道和4泳道均出現了明顯條帶,橫向比對預染Marker,各目的條帶的分子量大小正確。以Tubulin抗體免疫印跡后,1泳道和3泳道均出現了明顯條帶,橫向比對預染Marker,各目的條帶的分子量大小正確。該結果顯示,在COS7細胞中,CLIC1缺失第49~51位氨基酸后,其在細胞中的定位也發生明顯改變,即CLIC1在細胞質和細胞核中均有表達,表達量基本無差別,而CLIC1(Δ49-51)在細胞質和細胞中也均有表達,但細胞質中的表達量明顯高于細胞核。
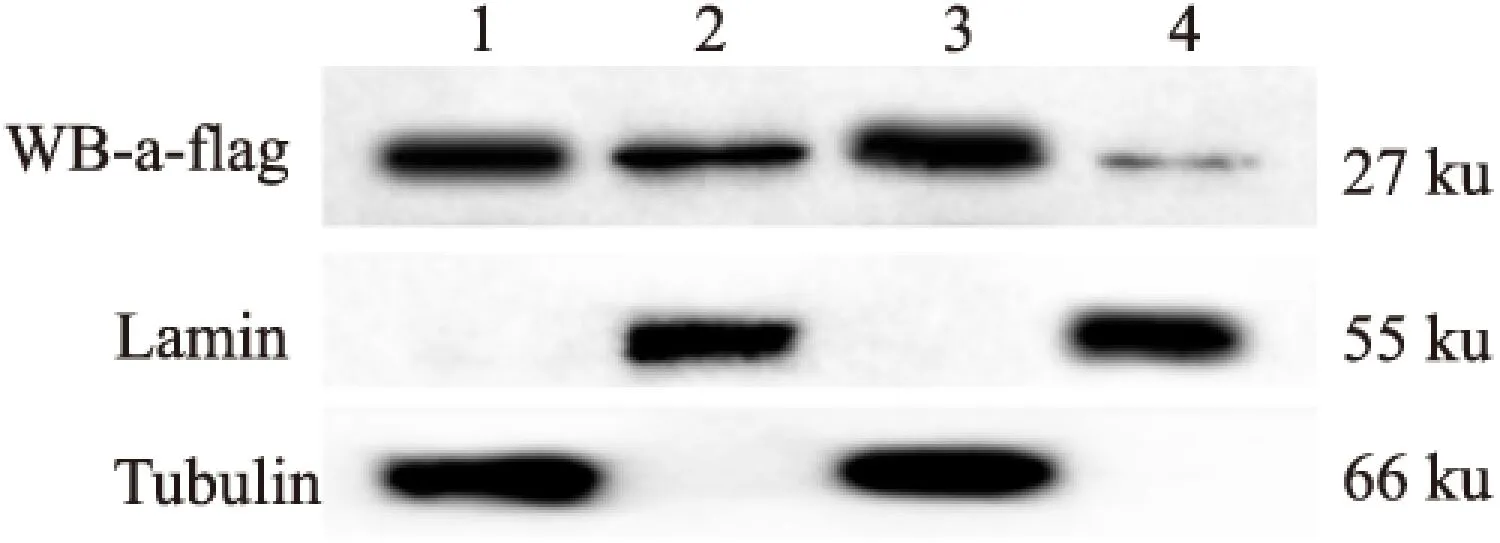
圖6 CLIC1、CLIC1(Δ49-51)的核質分離實驗
1:單轉CLIC1-FLAG分離的CLIC1細胞質的裂解液;2:單轉CLIC1-FLAG分離的CLIC1細胞核的裂解液;3:單轉CLIC1(Δ49-51)-FLAG分離的CLIC1(Δ49-51)細胞質的裂解液;4:單轉CLIC1(Δ49-51)-FLAG分離的CLIC1(Δ49-51)細胞核的裂解液
3 討論
CLIC1是真核細胞的重要陰離子通道。研究[9]表明,保守的陽離子基團能增強CLIC1的膜結合能力。把CLIC1跨膜結構域的氨基酸殘基第49位的賴氨酸、50位的精氨酸、51位的精氨酸分別用3個連續帶負電荷的谷氨酸代替后,即剔除KRR基序能夠阻礙膜結合,中斷靜電相互作用。相反,帶正電荷的KRR基序能夠形成帶正電的陽離子基團即正電元件,CLIC1的跨膜結構域綁定和插入該正電元件,能進一步增強CLIC1的跨膜和插入。被帶負電荷的氨基酸替代后,CLIC1蛋白的二級、三級、四級結構發生了改變,減少了整體螺旋度,增加Trp35的極性,進而會影響其功能。
本研究構建了CLIC1第49~51位3個氨基酸的缺失突變體CLIC1(Δ49-51),初步探索其對CLIC1蛋白功能的影響。首先把重組質粒pcDNA3.1-CLIC1-FLAG、pcDNA3.1-CLIC1(Δ49-51)-FLAG分別轉染至HEK 293T和COS7細胞中,比較CLIC1突變體相對于野生型的表達及定位改變;鑒于本課題組已經證實野生型CLIC1蛋白與Sedlin蛋白存在相互作用[10],利用Western blot、免疫熒光、GST pulldown及免疫共沉淀實驗,研究缺失突變體CLIC1(Δ49-51)是否與Sedlin蛋白存在體內、體外的相互作用。結果顯示,不同于CLIC1與Sedlin蛋白在COS7細胞中存在共定位[11],CLIC1(Δ49-51)與野生型CLIC1在細胞內定位、表達及與Sedlin相互作用方面均存在明顯的差異。本研究結果顯示,缺失突變體CLIC1(Δ49-51)中帶正電荷KRR基序的缺失,使其蛋白結構及活性位點的構象發生改變,以及該位點與周圍氨基酸化學鍵的改變,進而也一定程度上改變了蛋白質的功能(圖7)。
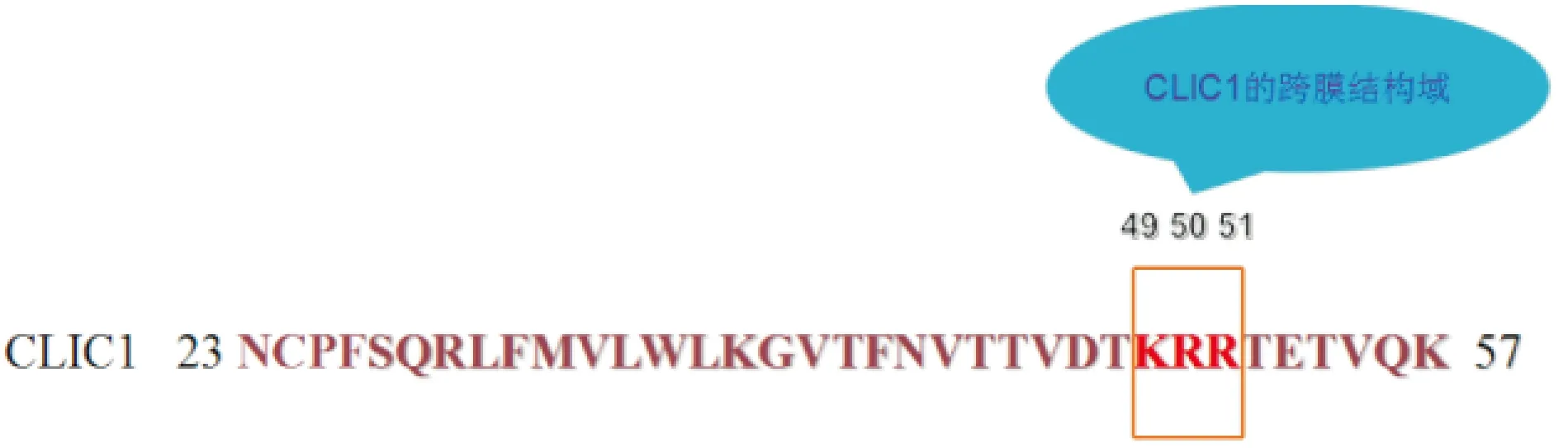
圖7 CLIC1氨基酸殘基(23~57)
CLIC1與腫瘤關系的研究比較多,有相關報道[12-14]表明CLIC1在乳腺癌、胃癌、大腸癌、肝癌中的表達明顯上調。CLIC1通過參與內質網到高爾基體的蛋白運輸進而影響其他調節蛋白的表達[15],參與腫瘤細胞的增殖、分化以及凋亡過程。因此,研究CLIC1的功能及關鍵氨基酸位點,為進一步探尋臨床腫瘤標志物或治療靶點提供重要依據。
