脊髓小腦性共濟失調3型患者家系分析
2018-11-15 08:15:22于青云吳衛文
中國臨床醫學 2018年5期
關鍵詞:癥狀
于青云,吳衛文,丁 晶*,汪 昕
1.復旦大學附屬中山醫院神經內科,上海 200032 2.復旦大學附屬中山醫院青浦分院神經內科,上海 201700
脊髓小腦性共濟失調(spinocerebellar ataxia,SCA)是常染色體顯性遺傳、以共濟失調為主要表現的一組疾病。SCA主要臨床表現為小腦性共濟失調癥狀,患者常可伴有錐體束受損、眼球運動障礙、周圍神經病變等臨床表現[1-2]。SCA主要依據患者典型的共濟失調主訴,結合頭顱MRI表現(小腦、腦干萎縮),排除可能累及腦干、小腦的其他變性疾病后確診[2]。
SCA目前分為40余種亞型,均具有特征性表現,但臨床上常易混淆。其中SCA3型又稱馬查多-約瑟夫疾病(Machado-Joseph disease,MJD),由Nakano等[3]首次報道。MJD是SCA亞型最常見的一種,在中國人群中占所有SCA患者的48%~49%[1]。目前確診SCA仍需要進行基因診斷[2],以明確不同亞型,指導進一步診療方案建立。多項研究[1,4]發現,ATXN3基因的CAG 重復序列發生擴增突變,導致該突變基因編碼產物多聚谷氨酰胺增多,并在神經組織內異常聚集,進而導致SCA3型的發生。本研究報告1家系SCA3型病例,并結合文獻總結本病的臨床表現及分型,旨在為臨床診治提供經驗。
1 資料與方法
1.1 病 史 先證者(Ⅲ 5)女性,28歲,以“進行性行走不穩2年余”為主訴于2016年10月至我院就診。2014年7月患者開始出現走路不穩,走路偶有跌倒,上下樓梯不受影響,不影響日常生活及工作。隨后患者自覺行走不穩癥狀逐漸加重,2015年6月(患者2014年10月懷孕,2015年6月29日分娩,無異常分娩史)分娩后自覺行走不穩較前明顯加重,上下樓梯須扶欄桿,同時出現說話不清,飲水嗆咳,視力較前下降,雙手持物不穩、笨拙、僵硬,雙下肢無明顯誘因淤斑。飲酒史10年余(多以紅酒為主,偶有白酒),否認金屬、毒物等異常物質接觸史。
1.2 體格及輔助檢查 入本院查體:神志清楚,言語稍含糊,雙眼水平凝視眼震(+)。抬頭肌力Ⅴ-級,雙上肢肌力Ⅴ-Ⅴ-Ⅴ-(Ⅴ-)級,雙下肢肌力Ⅴ級,雙上肢肌張力稍高、雙下肢肌張力增高;雙上肢腱反射(~),雙下肢腱反射(),髕陣攣(+)、踝陣攣(+),雙側Babinski征、Chaddock征(±),雙劃征(+)。雙側共濟運動完成差。閉目難立征(+),直線行走不能完成,剪刀步態。無脊柱后凸、高弓足。輔助檢查:血肌酸激酶(CK) 74 U/L、肌酸激酶同工酶(CK-MB)23 U/L、類風濕因子(RF)<9.750 0 U/mL、C反應蛋白(CRP)0.2 mg/L。頭顱MRI未見明顯異常。肌電圖示四肢體感誘發電位異常,未見明顯多發性周圍神經病變或肌源性損害的電生理改變。
2 結 果
2.1 基因檢測結果 SCA 3型基因三核苷酸CAG重復69次。在常染色體隱性遺傳痙攣性截癱15型相關基因ZFYVE26內含子區域發現1處雜合突變(圖1)。本例患者具有典型癥狀、體征、結合基因檢測結果明確診斷為SCA3型。

圖1 受試對象基因檢測結果(PCR+毛細管電泳)
A:先證者基因; B:先證者之女(Ⅳ6)基因;C:先證者之子(Ⅳ5)基因
2.2 家系調查 先證者家系4代中共有4人發病(圖2)。本家系先證者外祖父(Ⅰ1)發病年齡為45歲,第2代中Ⅱ2、Ⅱ3發病年齡分別為40歲、42歲;第3代中目前僅先證者發病,其發病年齡為26歲。
該家系基因檢測結果顯示:先證者的兩個姐姐及弟弟SCA 3型基因CAG重復次數在正常范圍內。先證者的兒子(Ⅳ5)CAG重復次數為67次,女兒(Ⅳ6)CAG重復次數為71次;其兒子及女兒因年齡較小,目前尚未出現明顯異常。
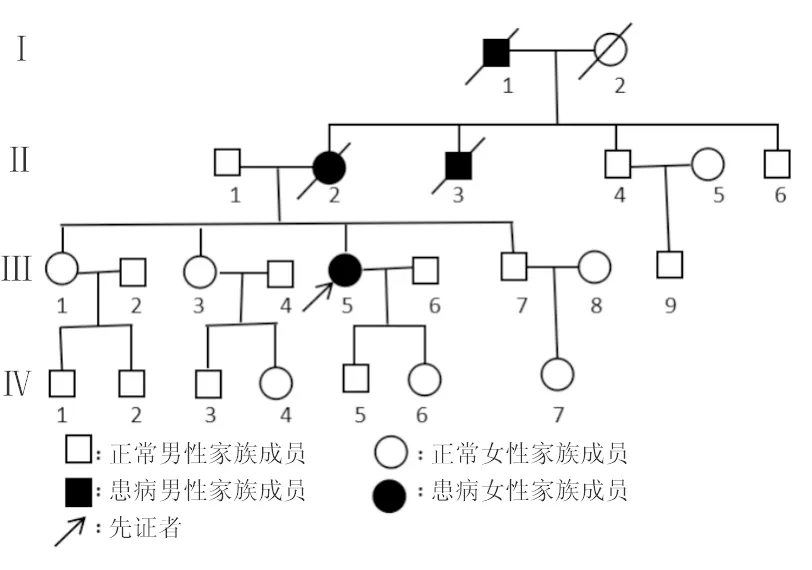
圖2 該例患者家系圖
患者外祖父Ⅰ1、母親Ⅱ2、舅舅Ⅱ3均已故,無法配合進行基因檢測
3 討 論
3.1 SCA分型 根據臨床癥狀體征、遺傳方式等,SCA可分為Ⅰ、Ⅱ、Ⅲ型。其中Ⅰ型包括SCA1~4、SCA8、SCA10、SCA12~23、SCA25、SCA27、SCA28、DRPLA,該型主要臨床表現為共濟失調癥狀,伴有眼球運動障礙、認知功能下降、其他錐體外系體征等;Ⅱ型即SCA7,該型臨床表現為共濟失調癥狀與色素性黃斑病變,且兩者具有相關性,同時可伴或不伴有眼肌麻痹癥狀、錐體外系體征;Ⅲ型包括SCA5、SCA6、SCA11、SCA26、SCA29、SCA30、SCA31,主要表現為單純共濟失調綜合征[5]。SCA3型在SCA各亞型中發病率最高[1]。
3.2 SCA3相關基因突變及蛋白 ATXN3基因CAG 的擴增突變是SCA3的基因突變形式。1993年,Takiyama等[6]首次發現,SCA3的致病基因位于染色體14q24.3-q32。1994年,日本Kawaguchi等[4]將其致病基因克隆到了染色體14q32.1,稱為ATXN3基因,也稱為MJD1基因。2001年,日本Ichikawa等[7]首次報道了ATXN3基因的結構,該基因的第10個外顯子上出現CAG的重復擴增,形成CAG重復序列。當ATXN3基因上的CAG重復序列出現異常擴增突變則可能致病。
ATXN3基因編碼的蛋白為ataxin-3蛋白,野生型ataxin-3蛋白相對分子質量小,在人體組織的胞質及胞核中有廣泛表達。野生型ataxin-3蛋白由361個氨基酸組成,其中Josephin結構域位于該蛋白的N端,而UIM結構域和polyQ肽鏈位于C端[8-10]。polyQ擴展突變型ataxin-3蛋白可發生β樣錯誤折疊,而發生錯誤折疊的ataxin-3蛋白可在其中樞神經元內形成泛素陽性核內包涵體,從而使突變個體發生SCA3[11-13]。2011年,德國Koch等[11]首次提出polyQ蛋白毒性片段學說,發現擴展突變型的ataxin-3蛋白中的polyQ蛋白多短小,且越短小polyQ片段致細胞毒性越強。而2012年 Costa等[14]也發現,SCA3患者病情與擴展突變型ataxin-3蛋白中polyQ毒性片段正相關,毒性片段越多,患者癥狀越重。
3.3 SCA3表現 小腦性共濟失調是SCA3的主要臨床表現,患者常同時伴有錐體束受損,如腱反射亢進、病理征陽性、痙攣步態等體征。SCA3患者黑質-紋狀體受損可出現帕金森樣癥狀、肌張力障礙、舞蹈樣癥狀等。SCA3患者亦可出現眼球運動障礙、周圍神經病變、情感障礙、嗅覺異常、構音不清、聽力下降等多種表現[1,15]。Bettencourt等[1]根據SCA3患者不同的臨床特征,將SCA3表型分為5型:Ⅰ型為Joseph型,主要臨床表現為小腦性共濟失調癥狀,可伴或不伴眼球運動障礙、錐體系及黑質-紋狀體受累的癥狀體征;Ⅱ型為Thomas型,患者癥狀多較輕,可伴有錐體外系及周圍神經病變,錐體束征不一定出現,患者錐體外系或周圍神經病變明顯時,可發展成Ⅰ型或Ⅲ型;Ⅲ型為Machado型,多伴周圍神經病變,錐體系、錐體外系癥狀不一定出現(合并時相對較輕);Ⅳ型為帕金森型,多伴有帕金森樣癥狀、輕度小腦癥狀、伴或不伴遠端感覺運動神經病,對左旋多巴治療反應較好;Ⅴ型為痙攣性截癱型,多表現為單純或復雜的痙攣性截癱癥狀,同時伴或不伴小腦共濟失調癥狀,但尚未得到廣泛認可。
3.4 本家系患者表現 本家系中先證者主要表現為走路不穩,進食、進水嗆咳,雙手持物不穩、笨拙、僵硬感,視力下降,言語含糊,伴有雙眼水平凝視眼震(+),雙下肢肌張力增高,四肢腱反射亢進,剪刀步態,閉目難立征(+);臨床主要表現為痙攣性截癱伴共濟失調癥狀,其分型考慮為Ⅴ型。而患者的外祖父、母親、大舅(均已故)主要表現為行走不穩、飲水嗆咳、言語不清、持物不穩等小腦性共濟失調癥狀。本家系第1代為患者外祖父,發病年齡45歲;第2代分別為40歲、42歲發病;第3代目前僅先證者,26歲發病;第4代兒子和女兒ATXN3基因CAG重復次數均超過正常范圍,目前尚未出現明顯異常。CAG重復序列在減數分裂中具有不穩定性,ATXN3基因子代的CAG 重復序列有擴增突變趨勢,且擴增次數越多,SCA發病年齡越小,即遺傳早現[2]。本家系調查顯示,患者發病年齡逐代提前,具有遺傳早現現象。
3.5 SCA3鑒別診斷 SCA3須與Friedreich型共濟失調、遺傳性痙攣性截癱、其他原因所致獲得性共濟失調等疾病相鑒別。本例患者發病年齡較晚,四肢腱反射亢進,深感覺存在,未見明顯弓形足和脊柱后側凸畸形,可排除Friedreich型共濟失調。患者否認毒物、重金屬等異常物質接觸史,無副腫瘤表現相關病史,體質量無下降,頭顱MRI無血管性疾病表現,可排除獲得性共濟失調。遺傳性痙攣性截癱臨床三聯征主要表現為:進行性加重的雙下肢肌張力增高、剪刀步態、肌無力,而本例患者主要表現為雙下肢肌張力增高、肌力正常、剪刀步態,不能排除遺傳性痙攣性截癱,而基因檢測結果示SCA 3,遺傳性痙攣性截癱相關基因為攜帶基因。
綜上所述,SCA3屬于遺傳性疾病,國際上目前尚無特異性治療方法,而對癥治療僅可緩解患者癥狀。SCA通過基因檢測明確臨床分型,能為其臨床診治及基因治療方法提供參考。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
