脊髓小腦性共濟失調一家系報道
2018-11-15 02:21:42重慶醫科大學附屬南川人民醫院神經內科重慶408400
現代醫藥衛生 2018年21期
關鍵詞:癥狀
陳 莉(重慶醫科大學附屬南川人民醫院神經內科,重慶408400)
脊髓小腦性共濟失調(SCA)是遺傳性共濟失調的主要類型,是一種起病隱匿、逐步進展、高度遺傳異質性神經系統變性病,主要累及人類中樞神經系統。成年起病、呈常染色體顯性遺傳及小腦性共濟失調為本病的共同特征。現報道本病有30余種亞型,我國最常見為SCA3型。現報道如下。
1 臨床資料
1.1 病例介紹 患者,女,40歲,主因“步態不穩、吐字不清3年”于2017年3月16日收入本院神經內科。主要表現:患者3年前無明顯誘因出現走路搖晃,步態不穩,伴頭暈、視物模糊,行走時向前沖,向后跌倒1次,伴雙上肢精細動作緩慢,講話緩慢,吐字不清,飲水嗆咳,無頭痛、嘔吐,無肢體抖動、身體發僵。2年前出現睡眠中大喊大叫,肢體舞動,噩夢,清醒時無幻覺。半年前出現便秘,伴記憶力明顯減退。病程中無情緒低落、肢體疼痛。既往體健,否認腦卒中、創傷史。家族史:患者的外婆、姨姥姥、舅舅、母親、二姨、舅舅的2個女兒、二姨的2個兒女均有類似癥狀,患者的兒子智力較同齡人稍差。
查體:右側臥位血壓 122∕84 mm Hg(1 mm Hg=0.133 kPa),心率 72 次∕分,立位血壓 115∕76 mm Hg,心率79次∕分,內科系統查體無陽性體征。神經系統查體:神志清楚,構音障礙,計算力、記憶力減退,反應遲鈍,顱神經查體無陽性體征。四肢肌容積正常,四肢肌力5級,四肢肌張力減低,雙側指鼻試驗、跟膝脛試驗欠穩準,輪替試驗動作幅度及速度減慢,閉目難立征陽性。行走時步基增寬,醉酒樣步態,后拉試驗陰性。雙側針刺覺及音叉振動覺對稱。四肢腱反射未引出。雙側Hoffmann征、Rosolimo征、掌頦反射、Babinskin征均陰性。輔助檢查:頭顱CT可見小腦萎縮(圖1)。三大常規、凝血分析、肝腎功能、電解質、血脂、血糖、葉酸、維生素B12、鐵蛋白、腫瘤標志物、抗O、類風濕因子、C反應蛋白、血細胞沉降率(血沉)、乙肝五項、糖化血紅蛋白、甲功全套、蛋白電泳、免疫全套化驗均正常。血清同型半胱氨酸 18.1 μmol∕L。胸部 X 線片、心電圖、腹部彩色多普勒超聲(彩超)、婦科超聲、泌尿系超聲、經胸心臟超聲心動圖均正常。黑質回聲強度Ⅱ級。眼科會診未見KF環,眼底照相正常。進一步完善頭顱磁共振成像(MRI)檢查仍可見小腦萎縮改變(圖2)。頸椎MRI檢查脊髓未見異常信號改變(圖3)。患者均成年起病,臨床以共濟失調癥狀體征為主,影像學檢查可見小腦萎縮,結合家系調查代代均發病,男女均患病,且發病年齡逐漸提前,具有遺傳早現表現,故臨床診斷為共濟失調綜合征:SCA可能性大。患者由于經濟原因未進一步行基因檢測,無法明確具體分型。告知患者及家屬病情后放棄治療回家休養,患者出院時癥狀無明顯緩解。3個月后隨訪,患者病情無明顯變化,6個月后隨訪,患者步態不穩加重,需在家人攙扶下緩慢行走,跌倒2次。目前仍在隨訪中。
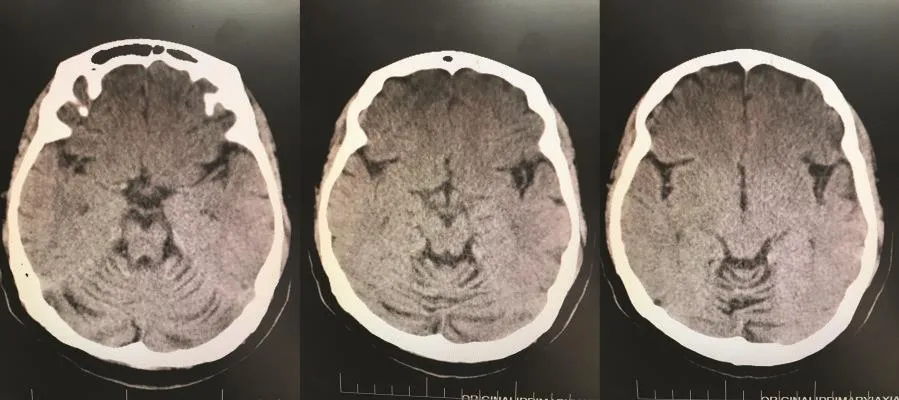
圖1 SCA先證者頭顱CT檢查示小腦蚓部和雙側小腦半球明顯萎縮
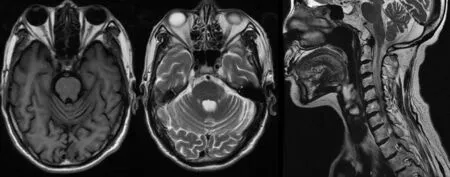
圖2 SCA先證者頭顱MRI檢查示小腦蚓部和雙側小腦半球萎縮
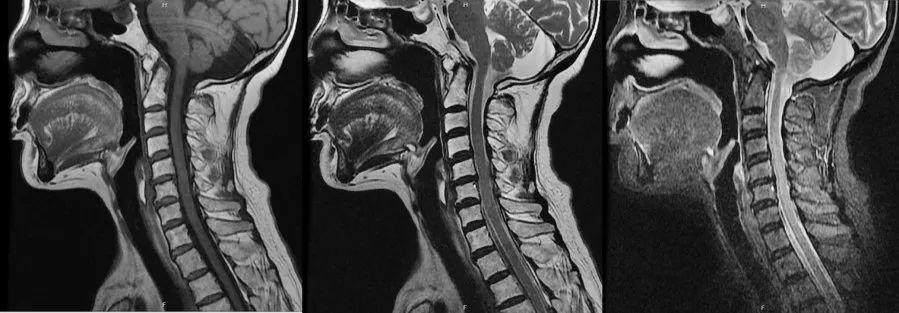
圖3 SCA先證者頸椎MRI檢查示頸髓未見異常信號改變
1.2 家系調查 該患者家系4代共29人,均無近親結婚,患病10例(男2例,女8例)。先證者的外婆(Ⅰ2,55歲發病,已故)、姨姥姥(Ⅰ1,50歲發病,已故)、舅舅(Ⅱ2,44歲發病,已故)、母親(Ⅱ5,54歲發病,已故)、二姨(Ⅱ3,48歲發病,已故)、舅舅的兩位女兒(Ⅲ1、Ⅲ2,分別于 42、39歲發病)、二姨的兩位兒女(Ⅲ6、Ⅲ7,分別于39、38歲發病)均有類似癥狀,患者的兒子智力較同齡人稍差,未發病。第Ⅰ代平均發病年齡52.5歲,第Ⅱ代48.67歲,第Ⅲ代39.6歲。第Ⅰ代和第Ⅱ代患者全部去世,第Ⅳ代目前未發病。SCA患者家系圖譜見圖4。
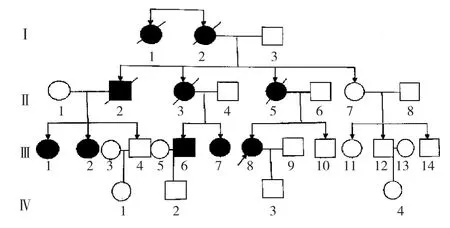
圖4 SCA患者家系圖譜
2 討 論
SCA是一大類由于遺傳因素造成的神經系統變性疾病,多為常染色體顯性遺傳,占神經系統遺傳性疾病的 10%~15%,患病率為(1∕10~4∕10)萬,各種族均可見[1]。共同臨床特征表現為成年起病、呈常染色體顯性遺傳及小腦性共濟失調等,具有在連續數代中發病年齡提前和病情加重的特征(遺傳早現現象)[2]。SCA有明顯的遺傳異質性和基因多效性,使患者臨床表現錯綜復雜[3]。目前,發現SCA1~29等若干亞型均為單基因遺傳病,已經發現30多種致病基因位點[4],我國常見的SCA亞型為SCA3,基因突變位點在14q24.3~q32,以小腦性共濟失調、錐體系和椎體外系癥狀、進行性眼外肌麻痹、遠端肌萎縮等為臨床特征[5]。
目前,認為SCA的發病機制可能與多聚谷氨酰胺擴增、非編碼區擴增、常規突變有關。由于SCA不同亞型基因突變位點的不同導致該病不同亞型損傷部位和發病機制不同,臨床表現也各異,在基因突變和臨床癥狀上既有重疊又有差異,稱之為SCA的多態性和異質性[6]。基因檢測是確定SCA基因亞型的唯一方法。
SCA以小腦、腦干和脊髓變性萎縮為共同的病理改變。大體臨床表現為:30~40歲隱匿起病,緩慢進展;首發癥狀多以雙下肢共濟失調為主,表現為走路搖晃、步基增寬或突然跌倒,伴有雙手笨拙及意向性震顫、辨距不良、構音障礙、眼球震顫等。查體可見肌張力障礙、錐體束征和深感覺障礙[7]。該病例家系中無近期結婚現象,男女均有發病,發病年齡38~55歲,以相似癥狀起病,后代較前代發病年齡逐漸減小,符合遺傳早現現象。SCA患者的CT或MRI可提示小腦和腦干萎縮。但SCA各亞型臨床表現大多相互重疊,發病初期影像學檢查可無異常,僅根據臨床表現和影像學檢查無法進行精確分型。基因檢測為確診SCA的“金標準”,但對于基層醫院大多數以農村患者為主,無條件完善基因檢測時仍以臨床診斷為主,需臨床醫生對該疾病提高認識。
目前,SCA尚無完全治愈的方法,臨床上以對癥藥物治療為主,目前研究較為熱門的治療方法為干細胞移植[8-9],但臨床效果欠佳,還有待進一步研究。SCA和其他神經系統變性病一致,其主要治療目的為減輕患者臨床癥狀、延緩疾病惡化速度、提高患者的生活質量[10]。
綜上所述,對于以共濟失調癥狀為首發癥狀就診的患者,臨床醫生必須詳細詢問病史,仔細對患者進行體格檢查,尤其是對于存在神經系統癥狀合并家族史的患者,應考慮到神經系統遺傳病的可能。在排除其他導致共濟失調的病因后,進行基因檢測有助于明確診斷[11]。同時,對SCA家族史中的健康人及早進行基因檢測,有助于預防和延緩疾病的發生、發展[12]。SCA患者及家族中健康人在結婚生育時鼓勵進行遺傳咨詢和產前篩查,盡可能地減少患兒出生[13],是優生優育、預防缺陷兒出生的重要手段。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
