藥用植物冬凌草高通量轉錄組測序與分析△
2019-01-16 00:59:16陳延清胡志剛劉大會黃必勝劉迪
中國現代中藥 2018年12期
陳延清,胡志剛,劉大會,黃必勝,劉迪
(湖北中醫藥大學 藥學院,湖北 武漢 430065)
大多數唇形科藥用植物味甘苦,性微寒,中國民間習用于治療失眠、消化不良、風濕病、腫瘤等[1-2]。現代化學和藥理研究表明,唇形科植物主要的生物活性成分為二萜化合物,由于其復雜多樣的碳骨架和具有多種抗腫瘤藥理活性成分,二萜類化合物已經成為抗癌藥物候選藥物[3-4]。冬凌草Isodonrubescens(Hemsl.)H.Hara屬于唇形科中藥用價值和經濟價值較為重要的藥用植物,其中全草中含量較高二萜類化合物冬凌草甲素、冬凌草乙素具有良好殺菌消炎、抗腫瘤、抗氧化等作用。目前,冬凌草中的二萜類化合物的研究與市場開發成為了熱點[5-6]。隨著對冬凌草資源與品質的研究,其道地性和種質資源的遺傳規律探索也逐漸深入[7]。采用現代化學分析技術,對冬凌草最佳采收期[8],以及對不同產地來源主要二萜活性成分冬凌草甲素和冬凌草乙素的含量差異進行了研究[9],并建立了不同產地冬凌草藥材的HPLC-PDA指紋圖譜特征圖譜[10],闡述冬凌草藥材的最佳采收時期和產地差異。利用隨機引物標記RAPD和ISSR[11],以及特異標記簡單序列重復SSR(simple sequence repeat,SSR)[12]等分子標記技術,探索了冬凌草遺傳多樣性規律,為篩選優質種質資源提供了豐富的遺傳基礎,探討藥材道地性的形成機制。
高通量測序(next generation sequencing,NGS)技術是對傳統測序的革命性改變,轉錄組學研究方法轉錄組測序(RNA-seq)揭示藥用植物遺傳育種和基因進化規律,其具有通量高、成本低、不受基因組序列信息限制等優點[13]。對于非模式生物而言,例如冬凌草,轉錄組測序更偏重基因編碼區域,與龐大的基因組信息相比,重復元件和富含GC的區域比較少,拼接相對容易[14],轉錄組測序針對物種轉錄本直接進行測序,為基因組序列信息有限的非模式生物提供可能。本研究利用 Illumina Hiseq 4000對藥用植物冬凌草藥用部位葉和莖進行了高通量轉錄組測序,建立冬凌草轉錄組數據庫,為深入研究冬凌草中冬凌草甲素等有效成分的生物合成途徑及其調控機制提供基礎,為藥用活性成分的合成代謝以及藥材道地性品質的形成提供生物學依據。高效地發現非模式物種的新基因和SSR位點的信息分析,也為開展分子標記輔助育種、基因工程技術選育創制新的藥用冬凌草優良品種奠定基礎,對優質中藥材的品種選育和規范化栽培生產具有重要的指導意義。
1 材料與方法
1.1 材料
本研究的冬凌草樣品來自于道地產區河南濟源[15],采于藥材最佳采收期[8],在同一生境下,選取冬凌草地上部分葉和莖為研究材料,迅速用液氮研磨,用于 RNA樣品提取。
1.2 總RNA的提取文庫、構建和轉錄組測序
用 TRIzol?Reagent、試劑盒Plant RNA Purification Reaget試劑盒(Invitrogen,美國)提取、純化葉與莖總RNA[16]。分別用Nanodrop 2000與 Agilent 2100 Bioanalyzer進行總RNA質量和純度檢測。為了獲得盡可能豐富的冬凌草采收期轉錄組信息,特別地將同珠的葉和莖的RNA混合。富集mRNA使用 Dynabeads Oligo(dT)25的磁珠。在Thermomixer中加入fragmentation buffer將mRNA打斷為短片段,添加0.1 mol·L-1Mg2+緩沖液到所得的 mRNA 中,高溫下使其片斷化后,然后將 mRNA為模板反轉合成cDNA、然后通過磁珠純化、末端修復、3’末端加堿基 A、加測序接頭等步驟后選擇片段大小,進行PCR擴增,adaptor構建冬凌草轉錄組測序文庫,使用Illumina HiSeq 4000 進行測序及分析。
1.3 測序數據質量處理及組裝
使用SeqPrep(https://github.com/jstjohn/SeqPrep)、Sickle(https://github.com/najoshi/sickle)軟件進行過濾原始數據,從而去除含有Primer/Adaptor的序列、含N比率超過10%的reads和含接頭的低質量reads,并且使Q30(堿基測序錯誤率低于0.1%)達到80%以上。從而獲得去掉接頭后的測序序列(cleanreads)進行后續分析。
由于缺乏冬凌草基因組的數據,因此利用Trinity軟件對冬凌草葉與莖轉錄組重頭組裝(de novo assembly),測序序列之間的重疊(overlap)信息組裝得到重疊群(contigs),依據兩端信息(paired-end)和重疊群的相似性,然后使用TGICL軟件進行聚類整合和延長得到非冗余的轉錄本(transcripts)。并從中選取最長的轉錄本作為非重復序列基因(unigenes)。Trinity 參數設置為K-mer=25,當序列延伸成重疊群時重復為 K-mer-1。拼接完畢后,將沒有匹配結果的短讀序的轉錄本刪除。處理與注釋冬凌草葉和莖的差異表達基因使用相同參考基因,得到同一時期冬凌草葉和莖所有Unigenes數據庫使用TGIGL的聚類組裝策略。
1.4 功能基因注釋
通過Transdecoder version v 2.0.1對unigenes的CDS進行了預測。利用Trinotate軟件包(v 2.0.0,https://trinotate.github.io/)的標準流程對All-unigenes進行了全面的注釋,用 Swissprot[17]、Pfam[18]、eggNOG[19]、Gene Ontology[20]、SignalP[21]、Rnammer[22]和KEGG Ontology數據庫注釋。
我們使用Trinotate軟件(版本:20140708)對預測出的編碼序列進行功能注釋。Trinotate軟件整合了多個數據庫,可以從不同角度對轉錄本進行功能注釋,使用軟件或數據庫如下:使用diamond(版本:0.5.3)blastx,SwissProt、TrEMBL和kegg作為數據庫,對轉錄本進行注釋;同時,還使用blastp(版本:2.2.28+),SwissProt作為數據庫,對轉錄本進行注釋;使用hmmscan(HMMER)軟件(版本:3.1),Pfam(Bateman et al.2002)作為數據庫,對預測的蛋白序列的結構域進行注釋;用SignalP(Petersen et al.2011)(版本:V4)軟件,預測信號肽;使用tmHMM(版本:2.0c)預測跨膜結構;使用RNAmmer(版本:1.2)預測rRNA基因。
1.5 基因表達水平及表達差異量分析
基于Trinity拼接得到的轉錄本,我們進行表達豐度計算。最先利用bowtie(版本:1.0.1;參數:mismatch=2)將每個樣品的reads分別比對到組裝出來的轉錄本上,得到bam格式的比對文件,然后使用RSEM(版本:v1.2.17)(http://deweylab.biostat.wisc.edu/rsem/)計算比對到轉錄本上的reads的表達量。為了便于不同基因之間進行表達量的比較,我們使用FPKM對表達量進行標準化。FPKM(expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced)[23]是每百萬fragments中來自某一基因每千個堿基長度的fragments數目,其同時考慮了測序深度和基因長度對fragments計數的影響。
差異表達基因(differentially expressed genes,DEGs)是基于基因表達水平分析中得到的read count數據,根據篩選閾值篩選出來的表達差異的基因。參考edgeR進行差異基因分析,篩選閾值選擇為False Discovery Rate(FDR)≤0.05,|log2FoldChange|≥1。其中FC(Fold Change)表示葉與莖間表達量的比值,其絕對值越大表明差異倍數越小,FDR值越大則為表達差異越不顯著[24]。使用軟件GOseq(版本:3.0)對樣品間差異表達基因進行GO庫注釋的分類統計和功能富集分析,使用KOBAS(http://ko-bas.cbi.pku.edu.cn/home.do)進行KEGG pathway富集分析,使用Fisher精確檢驗法對差異表達基因KEGG通路富集的顯著性進行評價。使用超幾何檢驗來找到差異表達基因中顯著富集的途徑。并經過多次檢測和校正后,定義P <0.05的通路途徑為顯著富集差異表達基因的途徑。
1.6 SSR分析
以冬凌草轉錄組為研究對象,使用MISA(Microsatellite)軟件進行SSR搜索。對Unigenes進行SSR檢測,參數設置為“1~12、2~6、3~5、4~5、5~4、6~4”,1~12代表單堿基重復至少12次才算SSR,雙堿基6次,三堿基5次,以此類推,重復單元最多有6個堿基。
2 結論與分析
2.1 總RNA質量檢測和組裝結果分析
經檢測,樣品總RNA的A260/A280均大于1.8,28 S/18 S在1.5~2.3之間,反應RNA完整性的RIN值在8.5~9.5之間為質量滿足建立和基因功能注釋、分類及樣本間異同性分析。
通過組裝,葉與莖冬凌草樣品共獲得3 7961個Unigene序列。All-Unigene總長為40366712 bp,平均長度為1063 bp,反應組裝效果的N50值為1810。其中N50是評估組裝質量最常用的指標之一,它反映了組裝結果的連續性和完整性。所得Unigene的數目隨長度的增加而減少,其Unigene大多集中在在200~3000 bp之間,平均Unigene為1063 bp。總的來說,Unigene的整體長度分布均勻(圖1)。

圖1 All-Unigene長度分布統計
2.2 Unigene蛋白數據庫比對結果
基于BLAST 算法對基因相似性分析,在37 961個Unigene 中,PFam庫中17842Unigene 同源匹配得到信息,在SignalP數據庫中有2394個,在eggNOG數據庫中有8769個,在 GO 數據庫中有18 776個,KEGG 數據庫中有23 936個(表1)。通常情況下測序的難度與基因組、轉錄組復雜程度有關。此外,由于缺乏參考基因組信息,當前研究非模式生物的轉錄組仍具有一定挑戰。即使目前國內對藥用植物的從頭轉錄組研究已有報道,但測序深度和基因檢出數目仍有待完善和提高[25-26]。
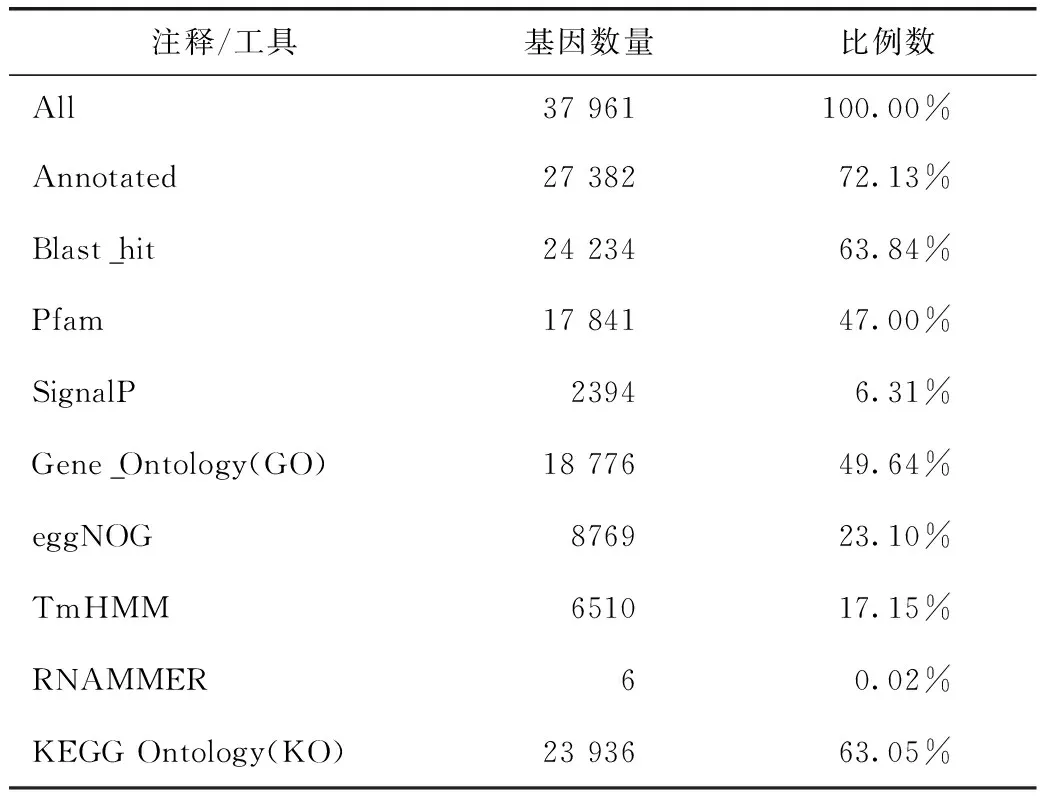
表1 All-Unigene蛋白數據庫統計結果
2.3 GO注釋和分類
GO注釋和分類是轉錄組數據研究中的一種國際標準化的基因功能分類手段,它提供了一套動態更新的標準詞匯表,用來全方面描述基因和基因產物的特性。GO功能分析由細胞組分、分子功能以及生物學過程三大部分組成。通過GO分析,37961Unigene中有18 775個獲得了GO注釋,被注釋為“細胞組分”有15 639個;16 455個被注釋為“分子功能”,1541515639被注釋為“生物學過程”。我們對其中的level2的GO注釋進行了統計(圖2)。
2.4 KEGG 注釋和分類
KEGG是全面分析生物體基因產物的代謝合成途徑以及產物功能的參考數據庫,根據生化途徑,提供基因的功能注釋和分類信息,對比分析表達基因在生物學上的系統行為。將6456個 Unigene 映射到 KEGG Pathway 數據庫中,注釋為代謝行為相關的序列共有4235個。其中涉及代謝(metabolism)、生物系統(organismal systems)環境信息處理(environmental information processing)、細胞進程(cellular processes)及遺傳信息處理(genetic information processing)5個大類及102條代謝通路,包含 Unigene 最多的是Ribosome(核糖體)共有389個,占糖酵解/糖異生(glycolysis/gluconeogenesis)共有 226個,占6%;其次是Oxidative phosphorylation(氧化磷酸化)共有244個,占3.8%,這些轉錄水平上的數據變化充分說明了冬凌草藥用部位在特定發育時期的新陳代謝變化(圖3)。進一步研究還發現,在代謝途徑中,與中藥有效活性成分生物合成相關Unigenes 序列分別是苯丙素類合成(phenylpropanoid biosynthesis)101條、萜類化合物骨架合成(terpenoid backbone biosynthesis)60 條、各種萜類化合物合成(sesquiterpenoid,triterpenoid,monoterpenoid and diterpenoid biosynthesis)6條、(莨菪類、哌啶類、吡啶類)生物堿合成(tropane,piperidine andpyridine alkaloid biosynthesis)15條、黃酮類成分合成(flavonoid,flavone,flavonol and isoflavonoid biosynthesis)30條(圖4)。
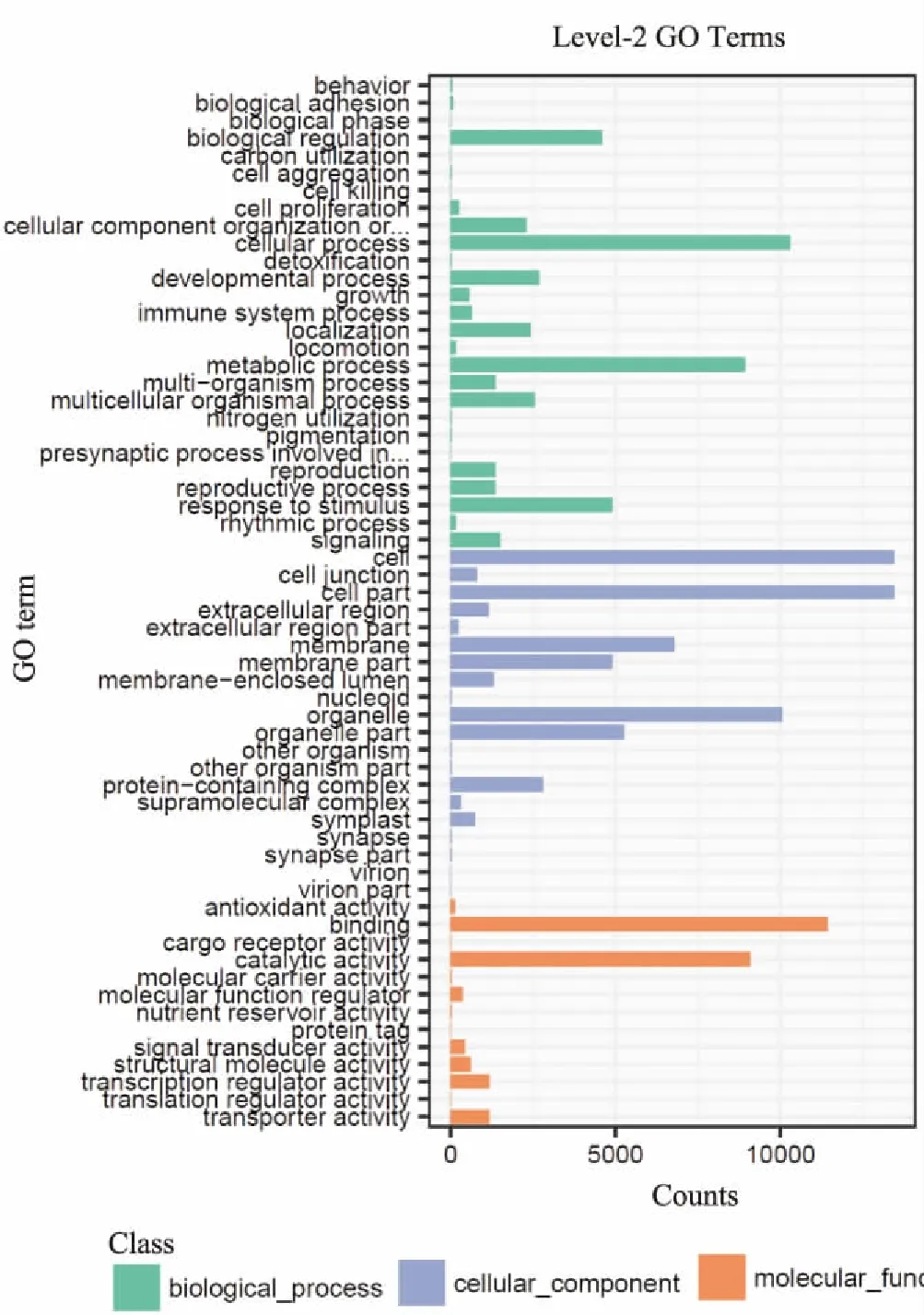
圖2 Level-2的GO注釋結果
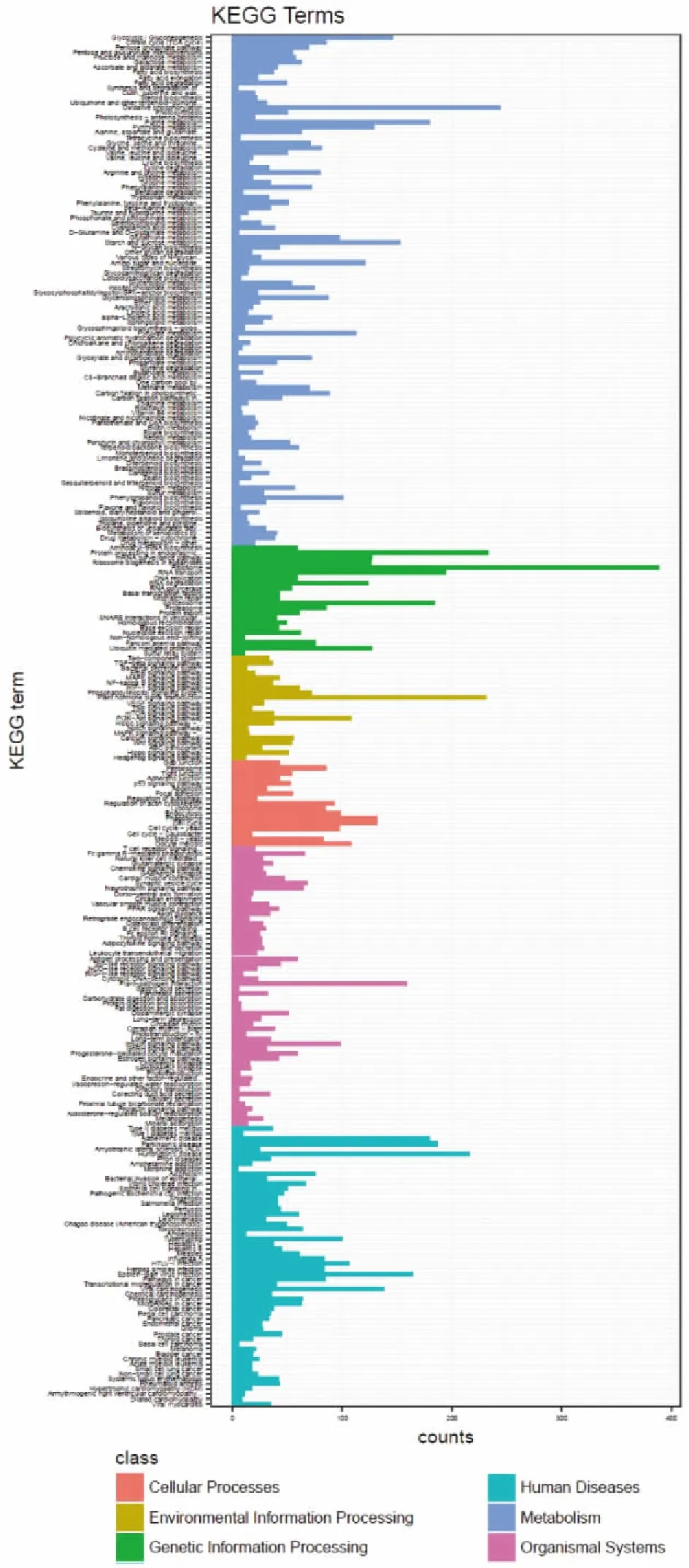
圖3 KEGG 注釋和分類結果
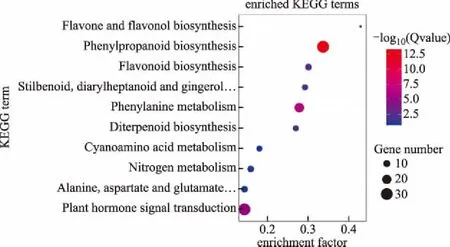
圖4 KEGG富集分析圖
2.5 二萜化合物合成途徑相關基因
植物二萜化合物主要合成途徑已經明確,分為三步:最先,活性異戊烯焦磷酸(IPP)及其異構體二甲基烯丙基焦磷酸(DMAPP)生成;之后為二萜化合物通用前體香葉基香葉基焦磷酸(GGPP)碳骨架的合成;最后在相關酶的作用下發生環化、羥基化等從而形成完整的二萜化合物分子[27]。利用本地化Blast的檢索方式在冬凌草中確定了本數據中與二萜化合物生物合成相關的候選基因。發現二萜合成途徑上的相關基因共有26個,其中與二萜骨架合成有關的基因有9個,其中與焦磷酸合成酶(copalyl diphosphate synthase)有關的 Unigene有6個,與貝殼杉烯合酶合成酶(KSL,kaurene synthase-like synthase)有關的unigene 有3個,具有細胞色素 P450(CYP450,cytochrome)功能的 Unigene共有5個。
2.6 差異表達基因的分析
通過冬凌草葉與莖兩個不同組織樣品的轉錄本/基因的表達量概率(FPKM scores)密度分布(圖5),不同樣品間差異基因在KEGG 通路富集具有特別顯著性差異(P< 0.01)的通路描述,可以鑒定出一些在葉和莖兩種組織在次生物質合成與代謝途徑中的差異表達基因。對冬凌草葉與莖兩種組織進行差異表達分析,得到差異表達基因4565個,其中在莖中表達較高的為1668個,在葉中表達較高的有2697個,與二萜類化合物合成相關的差異基因有15個。冬凌草葉與莖兩種組織差異基因表達圖如圖6。

圖5 冬凌草葉和莖基因表達量密度分布結果
2.7 SSR分析結果
簡單重復序列又稱微衛星序列(Simple Sequence Repeat,SSR)由于具有高度多態性,克服一般分子標記不確定、非特異的缺點,已在分子輔助育種、指紋數據庫的構建、品種純度鑒定及遺傳多樣性分析等方面得到廣泛應用[28-29]。在本次所測得的冬凌草轉錄組中,共檢測到8787條SSR序列,分布于7333條unigene中。SSR中二核苷酸重復的SSR標記有4016條,占比45.70%,是主要類型;其次是單核苷酸重復基序(2480條,占28.22%),接著是三核苷酸重復基序(1707條,占比19.43%)。

圖6 冬凌草葉與莖差異表達基因的MA圖和火山圖
3 討論
本文通過Illumina Hiseq4000 平臺,利用RNA-seq 技術對道地產地冬凌草的葉與莖進行了轉錄組測序與功能分析,初步建立了它們的基因表達圖譜,并通過比較藥用部位和非藥用部分合成途徑差異酶基因的發掘初步闡述了不同組織冬凌草成分差異的分子機制。并分析了轉錄組中的SSR,揭示了冬凌草中遺傳規律,也為藥用冬凌草的遺傳多樣性研究和分子標記開發奠定了分子基礎。
冬凌草葉和莖兩個組織樣品的高通量測序分析共獲得3 7961個 Unigene序列。大量Unigene在多個數據庫中不能匹配到已知基因,可能由于數據庫中由于缺少冬凌草基因組信息;通過對轉錄組數據的差異表達分析發現與冬凌草合成途徑有關的差異基因主要注釋在苯丙素類化合物、萜類化合物骨架合成、生物堿類化合物以及黃酮類成分生物合成途徑等通路中。冬凌草中的活性成分為二萜類物質[6],研究其生物合成途徑下游關鍵基因尤為重要。基于本文轉錄組數據,陳[30]等人成功克隆并初步驗證了下游IrCYP71功能基因,利用轉錄組數據挖掘藥用植物的功能基因已經成為重要手段[31]。對藥用植物的基因功能分化進行深入分析,并與活性成分種類、數量和積累變化相結合,進而從分子生藥學角度揭示由于不同生長時間和生長環境基因表達的差異與藥用部位中次生代謝成分差異的機制,才能為藥材質量控制與提高提供理論依據[32]。高通量轉錄組測序技術的應用同時也加速了SSR 位點的挖掘,本研究中冬凌草的 All-Unigenes 轉錄本中 SSR 出現的頻率特點為二核苷酸的重復單元類型最為豐富,這為后續冬凌草的種質資源和遺傳多樣性研究提供了豐富的分子標記數據。通過構建藥用植物生長發育的不同時期、不同器官和不同產地的轉錄組文庫,可以全方位地研究藥用植物中活性成份生物合成的關鍵酶基因的表達水平,從而有效地揭示次生代謝產物生物合成的途徑及其調控機制。
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
湖南教育·C版(2018年3期)2018-06-05 16:54:36
財經(2017年2期)2017-03-10 14:35:35
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
