納米氧化銅對水中三價砷的吸附特性及機理研究
2019-03-02 03:20:18韋安磊宋進喜
西北大學學報(自然科學版) 2019年1期
關(guān)鍵詞:模型
李 琦,高 歡,韋安磊,宋進喜
(西北大學 城市與環(huán)境學院,陜西 西安 710127)
砷是一種存在于地殼中的微量元素。水體中的砷一方面來自風化溶解等自然因素, 另一方面來源于人類工業(yè)活動, 比如冶金、 采礦、 制藥等產(chǎn)生的副產(chǎn)物[1]。 自然水體中砷通常以+3價或+5價的無機砷形態(tài)存在,無機砷為原生毒物,能抑制具有重要生理功能的多種酶的活力,長期接觸或飲用高濃度的含砷水體可導致多種癌癥發(fā)生,并且As(III)比As(V)毒性更強[2]。隨著全球工業(yè)的不斷發(fā)展,水體砷污染日益加劇。為了保護公眾用水安全,2001年,美國環(huán)保局(EPA)與美國國家科學院[3]聯(lián)合聲明對于水中砷濃度不應高于10μg/L。近年來,由于礦業(yè)活動的頻繁,我國14省20多個湖泊出現(xiàn)了嚴重的砷污染問題,砷檢出濃度高達1 900 μg/L[4]。此外,安建博等[5]調(diào)查發(fā)現(xiàn),西安市某縣5個自然村飲用水源長期存在超標現(xiàn)象,As(Ⅲ)含量高達(25.64±8.43)μg/L。
國內(nèi)外含砷廢水目前較為成熟的處理技術(shù)有混凝/沉淀法、離子交換法、生物膜法、吸附法等[6-8]。其中,吸附法由于不引入新的污染物,周期短等優(yōu)點處理含砷廢水更為理想。目前,被廣泛用于去除水體砷的吸附劑包括鋁,鐵,錳,鈦和磷酸鐵等[9-12]。由于在去除砷的過程中常規(guī)的吸附劑受到很多因素的限制,比如受水體pH影響、水中共存的多種競爭陰離子影響、吸附缺乏動力與選擇等,使得吸附有效性降低[13]。Reddy K J等[14]在實驗中偶然中發(fā)現(xiàn)氧化銅相比其他吸附劑有較好的吸附效率,并且不受溶液pH范圍、溶液中氧化還原電位及共存陰離子影響。
近年來,越來越多的研究開始將納米級的吸附材料應用于砷廢水的處理中。例如,錳納米顆粒、鐵納米顆粒、二氧化鈦納米顆粒、氧化銅納米顆粒等[15-17]。相比常規(guī)吸附劑,納米級吸附劑具有更大比表面積,吸附效率更高。
納米氧化銅由于其高效性和適應性強而受到廣泛關(guān)注, 雖然對于納米氧化銅吸附其他污染物質(zhì)的研究已經(jīng)有相關(guān)文獻報道, 但未有文獻對As(III)的吸附過程進行系統(tǒng)研究。本研究通過水熱法合成CuO-NPs作為吸附劑,探究CuO-NPs對As(III)的吸附效果。并且,結(jié)合透射電鏡(TEM),XRD(X射線衍射)和XPS(X射線光電子能譜)表征手段,分析吸附前后以及反應過程中可能的吸附機制,以期為CuO-NPs的應用和重金屬治理提供試驗依據(jù)。
1 材料與方法
1.1 試劑與儀器
試劑:乙酸銅(C4H6CuO·H2O)、硝酸(HNO3)、氫氧化鈉(NaOH)、無水乙醇(C2H5OH)均為分析純,購于廣州化學試劑廠;試驗中用水均為超純水。
儀器:透射電子顯微鏡(JEM-2100F,日本)、X-射線衍射儀(XRD-7000S/L,島津)、pH酸度計(pHS-3C,上海)、原子吸收分光光度計(AA-6300C,島津)、X射線光電子能譜儀/俄歇能譜儀(AXIS ULTRA; 英國)。
1.2 CuO-NPs的制備
水熱法制備CuO-NPs[18]。將2.5g的乙酸銅溶解于100mL的無水乙醇中,混合后滴入1.0g的氫氧化鈉,攪拌30min后移入聚四氟乙烯內(nèi)襯的高壓反應釜中,將反應釜置于120℃烘箱中恒溫2h,反應完成后冷卻至室溫,產(chǎn)物經(jīng)離心分離后,用無水乙醇及蒸餾水洗滌數(shù)次,然后將產(chǎn)物置于真空干燥箱恒溫80℃干燥24h。
1.3 吸附實驗
將0.1g的納米氧化銅加入裝有0.1L不同初始濃度的As(III)溶液的聚丙烯離心管中密封避光,于250r/min的轉(zhuǎn)速下依次振蕩一定時間,吸附溫度25°C,溶液初始pH值通過加入0.1mol/L HNO3和0.1mol/L NaOH調(diào)節(jié)至2~12。所有試驗均設(shè)置3次平行。
1.4 分析方法
1.4.1 CuO-NPs的表征 透射電子顯微鏡表征CuO-NP的形貌;X-射線衍射儀分析CuO-NPs的晶體結(jié)構(gòu),步長0.02°,掃描速度4°/min。
1.4.2 溶液中As濃度測定 采用GB-T 6730.67-2009測定吸附后的溶液As(III)的濃度。吸附量qe計算公式如下:
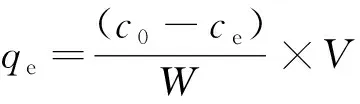
(1)
式中:C0和Ce為吸附前、 后吸附質(zhì)的濃度(μg/L),W為吸附劑的用量(g),V為溶液的體積(L)。
2 結(jié)果與討論
2.1 CuO-NPs的表征
由圖1可知,水熱法制備所得的CuO-NPs呈均勻的球形結(jié)構(gòu),平均粒徑在20~50nm,而且CuO-NPs顆粒沒有發(fā)生明顯的團聚,分散性良好。這表明水熱法制得的CuO-NPs顆粒具有較好的化學穩(wěn)定性。

圖1 CuO-NPs的TEM圖Fig.1 TEM image of CuO nanoparticles
圖2表明,在2θ=32.5°,35.6°,38.8°,48.7°,53.4°,58.3°,68.1°處出現(xiàn)尖銳的衍射峰,并且2θ=35.6°,38.8°處出現(xiàn)的衍射峰寬而強,在與氧化銅標準圖譜對照,屬于氧化銅的特征峰,說明實驗樣品為氧化銅,且結(jié)晶完整,為單斜晶系。根據(jù)衍射圖譜對應晶面所得數(shù)據(jù),選取最強的7個衍射峰(110,11-1,111,20-2,020,202,022),利用Scherrer公式進行晶粒粒徑估算,表達式如下:
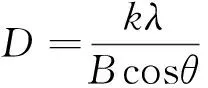
(2)
式中:D為粒徑;k為Scherrer常數(shù);k=0.89;λ為測量樣品時X射線波長(0.154 056nm);B為實測樣品衍射峰的半高寬度;θ為衍射角。計算得出CuO-NPs晶體晶粒粒徑在20~50nm,說明所形成的晶體晶粒尺寸為納米級別尺寸。
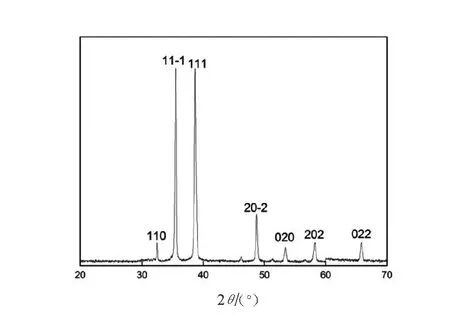
圖2 CuO-NPs的XRD圖Fig.2 XRD of CuO nanoparticles
零電荷點pHPZC是指材料表面正負電荷相等時的pH。由圖3可知,CuO-NPs的pHPZC為7.8。這說明,當溶液pH≤7.8時,CuO-NPs表面呈正電性,有利于溶液體系中陰離子的吸附去除;當溶液pH>7.8時,CuO-NPs表面呈負電性,有利于溶液體系中陽離子的吸附去除。
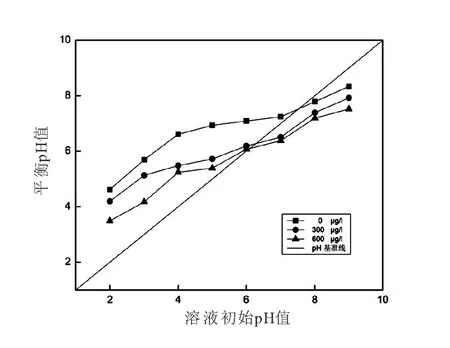
圖3 不同濃度As(III)溶液中的CuO-NPs的pHPZC變化Fig.3 The point of zero charge of CuO nanoparticles
2.2 pH對吸附As(III)的影響
pH是影響納米氧化銅去除水中As(III)的重要因素。實驗在As(III)初始濃度為600μg/L,CuO-NPs投加量為0.1g條件下,分析pH在3~11時的As(III)去除情況(圖4)。由圖4可知,在pH=3~8情況下,CuO-NPs對As(III)的去除率隨著pH升高而增加,在pH=8時去除率達到最大值97.05%。當pH≥9時,去除率隨著pH升高有所降低。
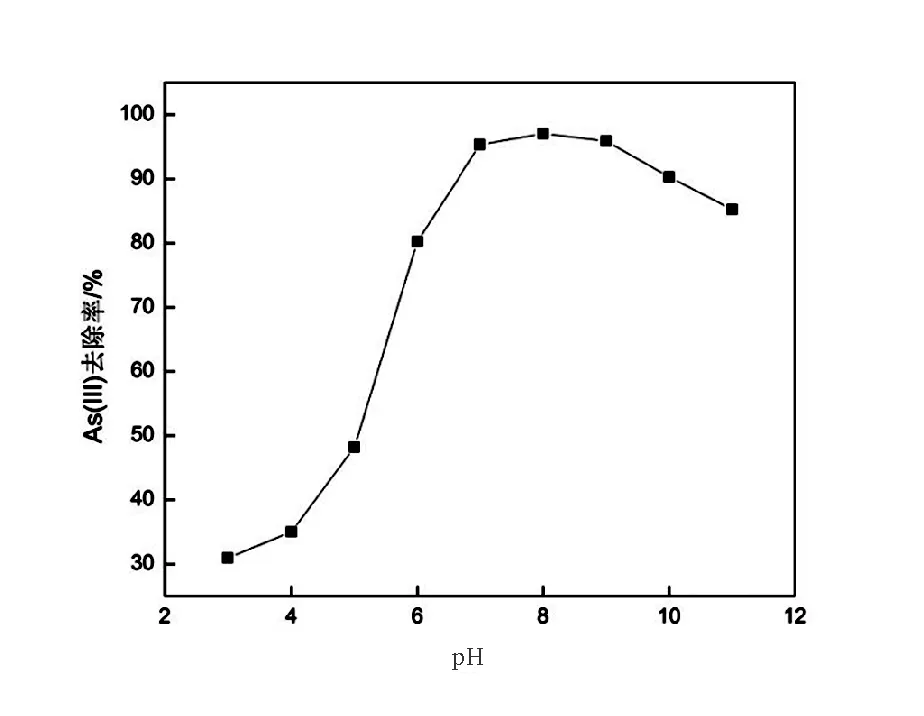
圖4 CuO-NPs在不同pH下對As(III)的去除率Fig.4 Effect of pH on the percentage adsorption of arsenic(III)
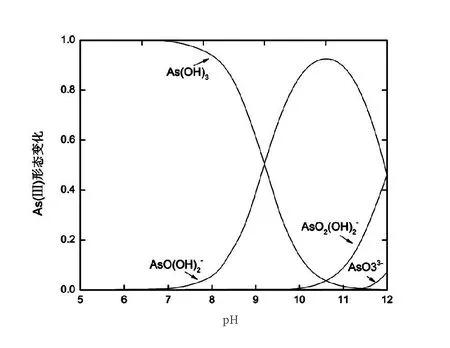
圖5 不同pH條件下As(III)的形態(tài)分布Fig.5 Morphological distribution of As (III) under different pH conditions
當溶液中存在300μg/L,600μg/L的As(III)時,CuO-NPs的pHPZC由7.8分別下降至6.5和6.1(圖3)。這表明CuO-NPs在吸附As(III)后,其表面電負性增強。通常而言,金屬氧化物表面羥基官能團的質(zhì)子化和脫質(zhì)子化過程決定了其pHPZC[18]。當金屬氧化物吸附某種物質(zhì)時,若僅形成外層絡(luò)合物,由于二者之間沒有發(fā)生化學反應,則其pHPZC不會被改變。那么,若pHPZC出現(xiàn)下降,則意味著金屬氧化物表面形成了帶負電的陰離子絡(luò)合物[19]。在本研究中,CuO-NPs吸附As(III)后,出現(xiàn)了pHPZC降低,這表明As(III)在CuO-NPs表面形成了帶負電的內(nèi)層絡(luò)合物。
2.3 吸附等溫模型
等溫吸附模型是通過對吸附過程中實驗數(shù)據(jù)的擬合來研究吸附劑對吸附質(zhì)的化學吸附行為。通過Langmuir方程(公式3)和Freundlich方程(公式4)來對CuO-NPs對As(III)吸附實驗數(shù)據(jù)進行擬合。其表達式如下所示:
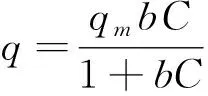
(3)
q=kFC1/n
(4)
式中:q為As(III)平衡吸附容量,μg/g;qmax為單層飽和吸附量,μg/g;C為吸附平衡時溶液中砷離子濃度,μg/L;b為Langmuir常數(shù),L/μg;KF和n為是Freundlich吸附平衡常數(shù)。
CuO-NPs吸附As(III)的等溫吸附模型擬合圖及擬合參數(shù)值如圖6,表1所示。可以看出,等溫吸附實驗結(jié)果與Langmuir等溫模型擬合度高。表1中Langmuir等溫方程擬合后的線性相關(guān)系數(shù)為0.999 8,高于采用Freundlich等溫方程擬合的線性相關(guān)系數(shù)0.957 0,由此說明Langmuir方程更適合描述CuO-NPs對As(III)的吸附過程。
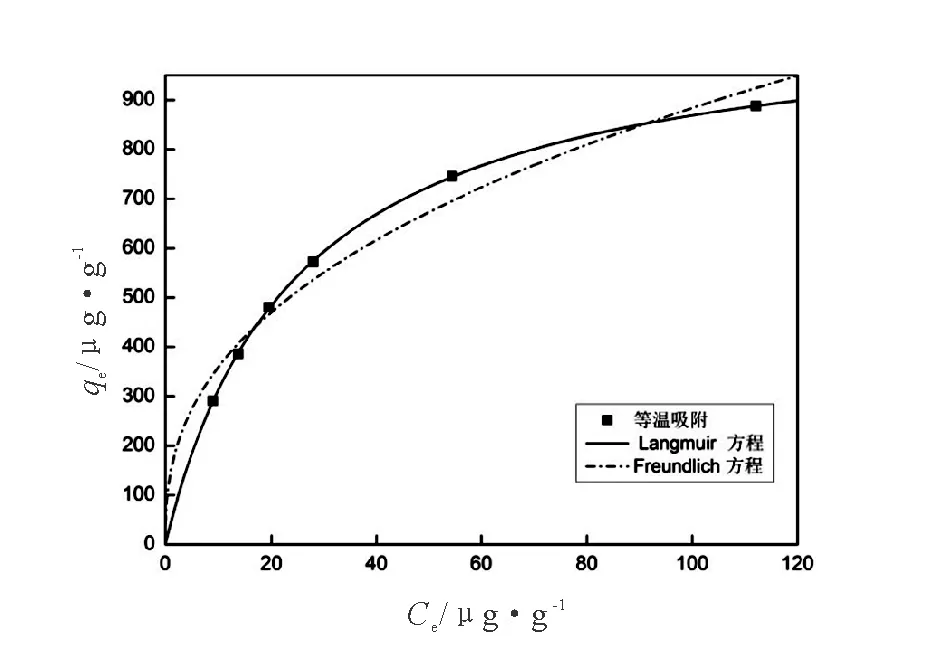
圖6 CuO-NPs吸附As(III)的等溫吸附擬合圖Fig.6 Equilibrium isotherm model for arsenic(III) adsorption
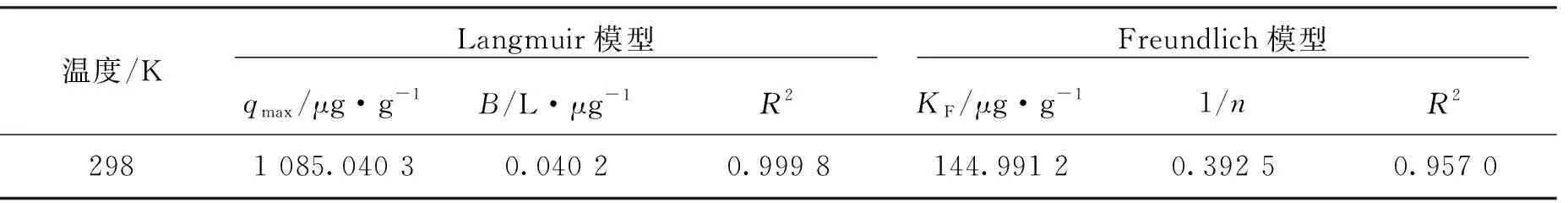
溫度/K Langmuir模型 Freundlich模型 qmax/μg·g-1B/L·μg-1R2KF/μg·g-11/nR22981 085.040 30.040 20.999 8144.991 20.392 50.957 0
此外,在Langmuir吸附等溫方程中,可定義一個無量綱的分離因子RL,它表示吸附過程的性質(zhì)。表達式為
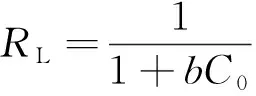
(5)
RL=0非可逆吸附;0
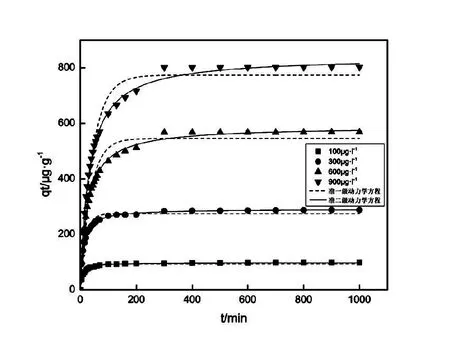
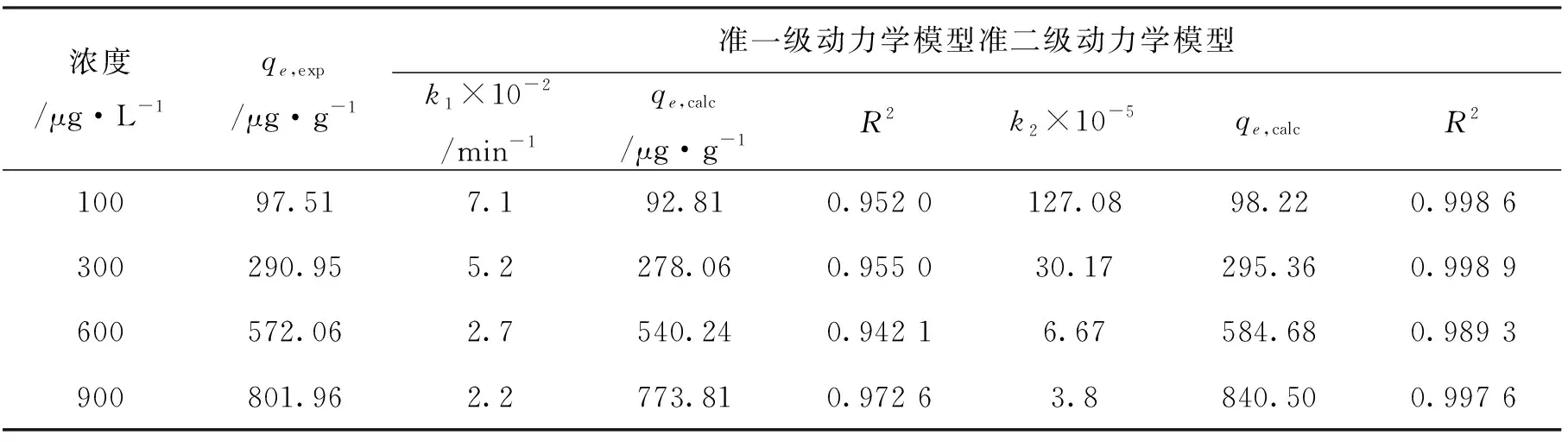
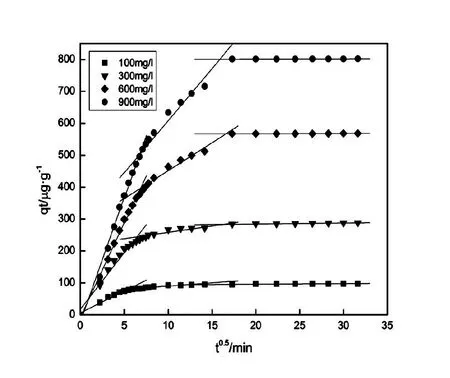
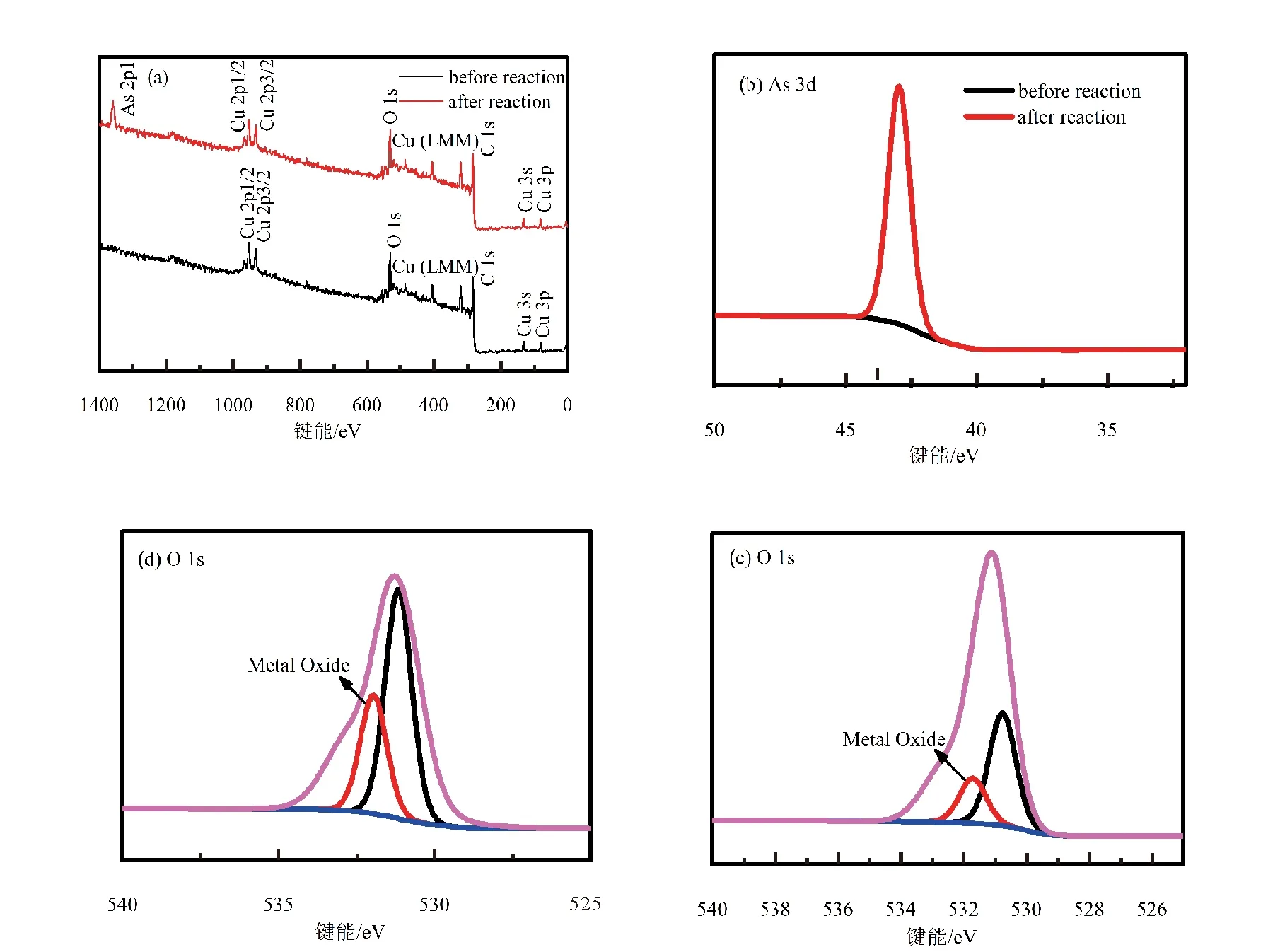
本次實驗中分離因子0 陳云嫩等[20]采用巰基乙酸改性麥糟吸附水中的As(III),最大吸附容量為252.300 μg/g;Sathishkumar等[21]用Aspergillus fumigatus菌體吸附去除As(III),發(fā)現(xiàn)最大吸附容量為538.900 0 μg/g;相比而言,本研究的最大吸附容量為1 085.040 3 μg/g,這說明納米氧化銅具有較高的吸附特性。 CuO-NPs對As(III)的吸附動力學曲線如圖7所示。 由圖可知, CuO-NPs對As(III)的吸附在前100min內(nèi)速率較快, 100min后速率逐漸減慢, 400min后趨于平衡。 此外, As(III)初始濃度越高, 吸附達到平衡需要的時間越長。 這是由于CuO-NPs表面的吸附點位是一定的[18], 隨著As(III)濃度的增加,需要投加更多量的CuO-NPs,提供充足的吸附點位來滿足As(III)的吸附,吸附達到平衡時間也就越長。 對CuO-NPs吸附As(III)的吸附動力學數(shù)據(jù)采用準一級動力學模型式(6)、準二級動力學模型式(7)及粒內(nèi)擴散模型式(8)進行擬合。表達式如下: qt=qe(1-e-k1t) (6) (7) qt=kpt0.5+C (8) 式中:qe和q2為準一級動力學模型和準二級動力學模型的平衡吸附量,mg/g;t為吸附時間,min;qt為t時刻的吸附量,mg/g;k1為準一級反應速率常數(shù)/min-1;k2為準二級反應速率常數(shù),g/(μg·min)-1;kP是顆粒內(nèi)擴散速率常數(shù),mg/(g·min0.5);C是吸附常數(shù)。 圖7 CuO-NPs吸附As(III)的吸附動力學Fig.7 Effect of contact time on arsenic(III) adsorption 準一級及準二級動力學模型擬合參數(shù)值列于表2。可以看出,準二級動力學模型計算得到的不同As(III)濃度下平衡吸附量和實驗數(shù)據(jù)更為相近,模型的相關(guān)系數(shù)皆大于準一級動力學模型。由圖7可看出,準二級動力學模型擬合程度更好。因此,準二級動力學模型能更好的描述CuO-NPs吸附As(III)的吸附過程。這說明該吸附過程是化學吸附過程,包括多個反應機制的過程,如As(Ⅲ)在溶液體相或界面處的擴散、表面的活化與去活化作用等[19]。隨著As(III)濃度的增加,準二級反應速率常數(shù)K2不斷減小,表明濃度越高,吸附達到平衡時的反應速率越小。 表2 CuO-NPs吸附As(III)的動力學參數(shù)值Tab.2 Kinetics parameters of the pseudo-first-order model and the pseudo-second-order model for arsenic(III) adsorption 圖8為As(III)在CuO-NPs的粒內(nèi)擴散模型圖。粒內(nèi)擴散模型圖呈現(xiàn)兩種結(jié)果,一種是內(nèi)擴散模型圖為一條直線,此時吸附過程受粒子內(nèi)擴散控制,若這條直線通過原點說明粒子內(nèi)擴散為唯一的決速步驟。另一種模型圖呈現(xiàn)多段線性,說明吸附過程復雜且受多個決速步驟控制。 圖8 CuO-NPs對As(III)的粒內(nèi)擴散模型Fig.8 The intra particle diffusion model for arsenic(III) adsorption 由圖8可以看出,吸附過程為三段線性,表明吸附過程復雜。第一階段As(III)吸附量隨t0.5不斷增加,說明As(III)在CuO-NPs上進行邊界層擴散;第二階段相比第一階段趨勢線的斜率變小,吸附量增加變慢,這是受到顆粒內(nèi)擴散的影響;第三階段的斜率幾乎為零,說明此時吸附趨于飽和,吸附達到平衡狀態(tài)。 為研究吸附過程中的熱效應,實驗采用吸附熱力學公式得出吸附焓ΔH,吸附熵ΔS及不同溫度下的吸附自由能ΔG。吸附熱力學公式如下: (9) (10) ΔG=-RTlnKd (11) 式中:R為氣體常數(shù),8.314J/(mol·K),b為Langmuir 吸附常數(shù)。 從表3可以看出,不同溫度下ΔG均為負值,說明CuO-NPs對As(III)的吸附過程是一個自發(fā)的過程,且溫度越高自發(fā)的程度越大。ΔH值和ΔS值分別為146.207 1KJ/mol,0.529 3J/(mol·K)。焓變ΔH為正,表明反應是吸熱反應。熵變ΔS為正,說明吸附過程在固液界面的無序性增加。 表3 CuO-NPs吸附As(III)的熱力學參數(shù)值Tab.3 Thermodynamic parameters for arsenic(III) adsorption 溶液pH=8時,CuO-NPs對As(III)的去除率達到最大,這主要是由于As(OH)3與納米氧化銅水合物之間發(fā)生了較強的配位交換(式(11))。當溶液呈酸性時,As(III)主要以As(OH)3形態(tài)存在(圖5),呈電中性,而此時CuO-NPs對As(III)的去除率較低。這主要是由于酸性條件較強導致的CuO-NPs表面質(zhì)子化不利于配位交換的發(fā)生。當溶液pH>8時,形態(tài)為As(OH)2O-的As(III)逐漸增多,此時As(III)的去除則逐漸以As(OH)2O-與CuO-NPs水合物之間的配位交換為主(式(13))。然而,由于CuO-NPs的零電荷點為7.8,隨著溶液堿性的增強,吸附劑表面負電性逐漸增大。這使得CuO-NPs與As(OH)2O-之間靜電排斥逐漸增強,因此,pH>8時,CuO-NPs對As(III)的去除率逐漸降低。 >Cu—OH+As(OH)3→—Cu—OH—As(OH)2 (12) >Cu—OH+As(OH)2O-→—Cu—OH—As(OH)O- (13) 圖9 反應前、后樣品XPS譜圖Fig.9 XPS spectra before and after the reaction 圖9為吸附As(III)前、后的CuO-NPs樣品的XPS圖譜。吸附過程結(jié)束后,吸附材料的全譜分析CuO-NPs表面存在As(圖9(a)),而且結(jié)合能在43.1 eV處的峰值進一步證實了As(III)被CuO-NPs吸附(圖9(b))。圖9(c)和(d)為吸附As(III)前后材料表面的O 1s高分辨率XPS譜線的分峰圖。吸附As(III)后,結(jié)合能在529.0~530.0 eV處的分峰增強(圖9(c)),這表明CuO-NPs表面金屬氧化物中的氧含量相對增加。此外,結(jié)合能在531.5~532.0 eV處的分峰也略微增強圖(9(d))。這可能是As(III)氧化態(tài)水合物樣品與空氣中二氧化碳作用所致。以上分析表明,吸附前后CuO-NPs樣品表面金屬氧化物所含的O化學鍵發(fā)生了顯著變化,吸附過程發(fā)生了化學吸附。這也與式(2)和(3)所描述的配位交換作用相一致。 1)采用水熱法成功制備了化學穩(wěn)定性較好、平均粒徑為20~50nm的CuO-NPs。 2)弱堿性條件有利于CuO-NPs吸附As(III);溶液pH=8時,CuO-NPs對As(III)的去除率最高,為97.05%。 3)As(III)在CuO-NPs上的等溫吸附符合Langmuir方程,最大吸附容量可達1 085.040 3 μg/g,且吸附過程自發(fā)吸熱、且符合準二級動力學方程。 4)CuO-NPs對As(III)的吸附,主要是通過氧化銅水合物與溶液中的As(OH)3和As(OH)2O-之間發(fā)生配位交換而實現(xiàn)。2.4 吸附動力學模型
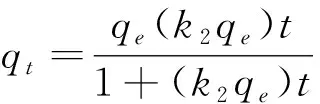
2.5 吸附熱力學模型
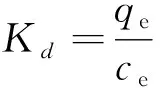
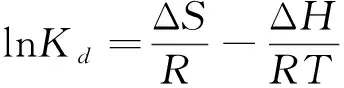

2.6 吸附機制
3 結(jié) 論
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網(wǎng)絡(luò)安全與數(shù)據(jù)管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數(shù)理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數(shù)學備考)(2021年9期)2021-11-24 01:14:36
成都醫(yī)學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數(shù)學備考)(2020年9期)2021-01-04 00:25:14
中學生數(shù)理化·七年級數(shù)學人教版(2020年10期)2020-11-26 08:24:50
數(shù)學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
