海洋動物蛋白藥物及其研究進展
2019-03-04 06:48:36劉智禹
漁業研究 2019年1期
潘 南,劉智禹
(1.福建省水產研究所,福建省海洋生物增養殖與高值化利用重點實驗室,福建 廈門 361013; 2.福建省海洋生物資源開發利用協同創新中心,福建 廈門 361013)
新藥研究開發核心之一是將基礎化學、生物分子和疾病聯系起來,完成高通量篩選、合成優化、藥理、毒理等臨床前試驗,主要由政府提供資金的科研院校、研究機構完成;其二是臨床研究、注冊上市和售后監督等,耗資大、耗時長、參與人數多,一般由制藥企業和委托研究機構(Contract research organization,CRO)完成。市場競爭的加劇促使制藥企業加快產品管線的更新換代,因此制藥產業界從本質上對藥效突出、技術前沿、開發前景好的基礎科研項目趨之若鶩。海洋動物經歷數百萬年的進化微調而演化出極高的靶向親和效力和穩定性的代謝產物[1],這使得它們成為制藥業藥品開發管線的靈感源泉之一。本文對海洋動物蛋白藥物及其研究現狀進行了梳理,總結了海兔毒素、芋螺毒素、海鞘毒素、海綿活性多肽、海葵活性多肽及其結構類似物、衍生物的來源、結構、藥理、分子靶標、臨床試驗研究現狀和發展瓶頸。
1 海兔毒素及其衍生物
1.1 海兔毒素Dolastatins
Pettit課題組于1987年首次報道了印度洋無殼軟體動物截尾海兔(Dolabellaauricularia)消化腺毒素dolastatins的分離、鑒定[2]。Dolastatins是一種線性五肽,包含dolavaline(Dov)、valine(Val)、dolaisoleuine(Dil)、dolaproine(Dap)和dolaphemine(Doe)五個子單元[3]。其中dolastatin 10和dolastatin 15在體外實驗中表現出極強的抑制細胞增殖、抗腫瘤活性,其通過與β微管蛋白結合,迫使細胞分裂停滯,進而凋亡。由于天然存在的dolastatins含量非常低,Pettit和合作者被迫開發了全新的化學合成方法,以便獲得足夠的材料進行基本的細胞生物學研究,在此基礎上進一步合成出一系列dolastatin衍生物auristatin PE(又稱Soblidotin;TZT-1027;YHI-501)、tasidotin(又稱Synthadotin;BSF 223651;ILX 651;LU 223651;SYN-D)和Cemadotin(又稱LU-103793;NSC D 669356)。然而由于藥物細胞毒性和/或缺乏功效,海兔毒素dolastatin 10、15衍生物均沒有進行超過臨床二期的研究(表1)[4]。
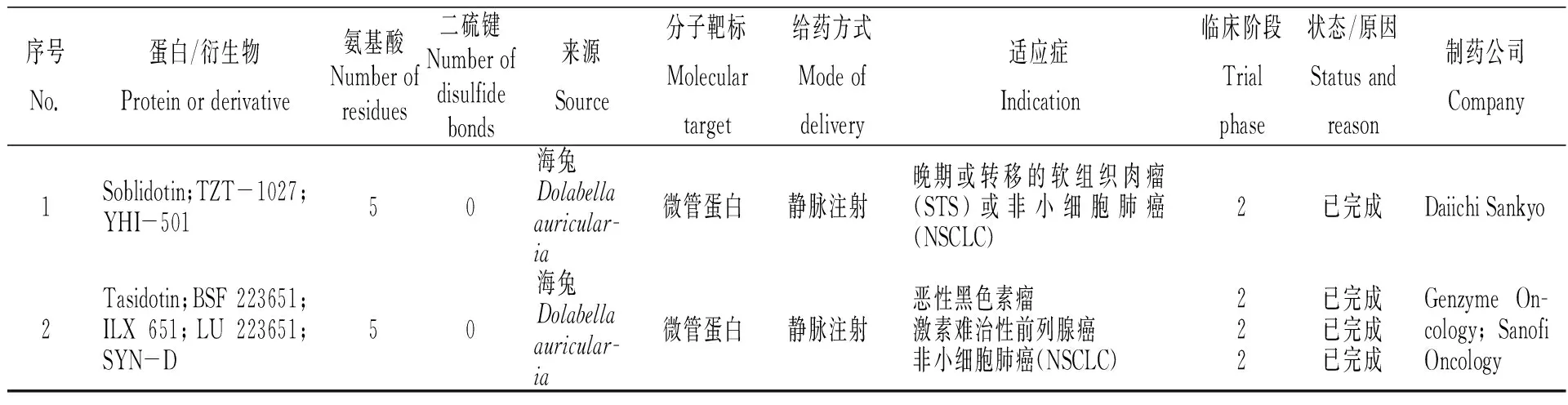
表1 經藥品監督管理機構審批進入臨床的海兔毒素dolastatin 10、15衍生物[4]
1.2 海兔毒素衍生物Auristatins
為了提高作用效力,Pettit課題組對dolastatin 10的C 端或N 端進行修飾,合成了auristatins 系列藥物(auristatin E、auristatin PHE 、auristatin PYE、auristatin-2-AQ、auristatin-6-AQ)[5],增強了藥物水溶性的同時仍然保持了顯著體外抗腫瘤效力(10~100 pM,取決于所用的細胞株系)。然而體內試驗結果仍然不盡理想。
1.3 經批準上市的auristatins 抗體-藥物偶合物(Adcetris?)
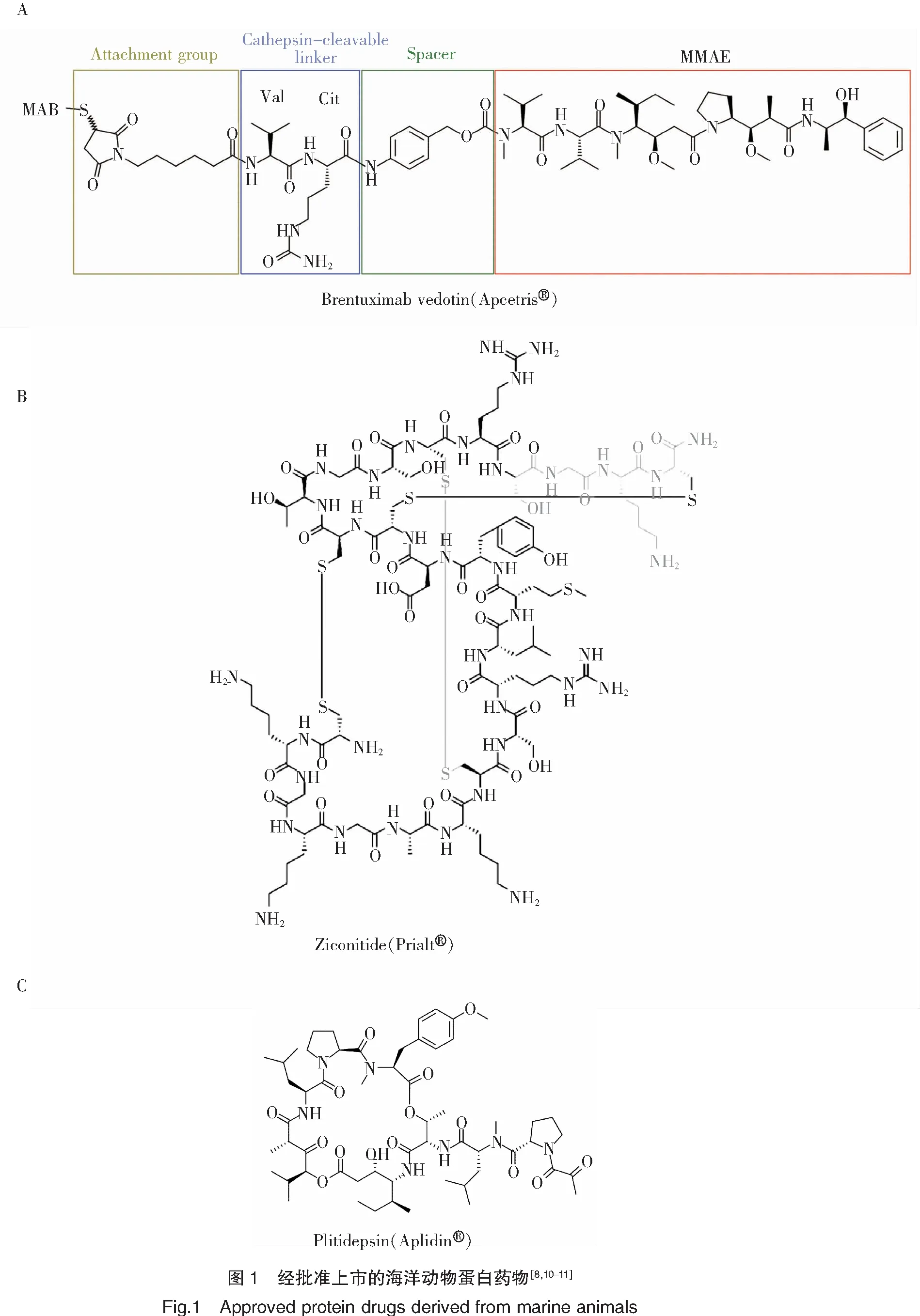
美國西雅圖遺傳學公司(Seattle Genetics)于2003年首次發表了其在auristatins 抗體-藥物偶合物的研究進展,其在單甲基auristatin E(MMAE)的N端通過對氨基芐氧羰基(PABC)間隔體鍵合纈氨酸-瓜氨酸(vc)連接體以偶合cAC10(CD30 抗體),形成特異性腫瘤靶向抗體-藥物偶合物Brentuximab vedotin(又稱cAC10-vcMMAE;SGN-35)(表2)[6]。通過cAC10 靶向識別腫瘤細胞膜上的CD30蛋白, Brentuximab vedotin與 CD30蛋白結合后經細胞網格蛋白的調節被內吞入細胞,在溶酶體中被蛋白酶水解釋放出MMAE,進一步抑制微管蛋白聚合,使細胞停留在G2-M期,促使細胞凋亡[7]。經歷一系列臨床研究,美國食品藥品管理局(FDA)于2011年、歐洲醫藥管理局(EMA)于2012年批準Brentuximab vedotin以商品名Adcetris?(圖1-A)上市,用于治療CD30表達的霍奇金淋巴瘤和系統性間變性大細胞淋巴瘤[8]。美國西雅圖遺傳學公司與日本武田公司(Millenium,Takeda)合作,該藥物于2014年在日本獲得衛生、勞動和福利部(MHLW)批準上市[9]。
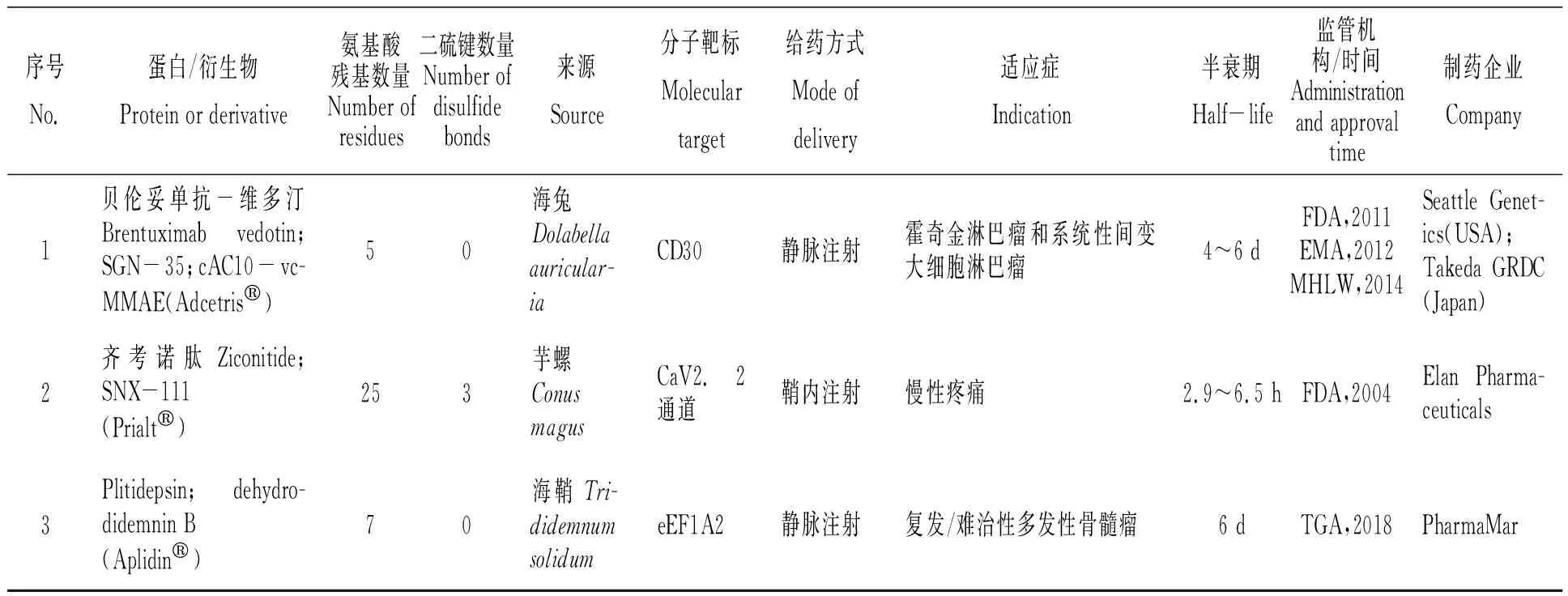
表2 經藥品監督管理機構批準上市的海洋動物蛋白藥物[6,8-11]
1.4 經審批進入臨床試驗的auristatins抗體-藥物偶合物
隨著腫瘤細胞靶點單克隆抗體研究和連接體技術的高速發展,為尋求更好的臨床試驗結果,高效、高活性的海洋動物細胞毒性蛋白受到單克隆抗體藥物研究者的青睞。以海兔毒素dolastatin 10為母體化合物合成的auristatins系列被廣泛應用于抗體-藥物偶合物臨床試驗中。表3總結了目前經審批進入臨床試驗的auristatins抗體-藥物偶合物,但必須指出的是,這份表格不是“完整的”,原因一是表格內容采用已公開的科學研究報道(論文、專利)[12-34]和公共數據庫(NIH ClinicalTrial.gov數據庫、NIH PubChem公共化學數據庫、Adis Insight Springer數據庫)資料[35-37],臨床試驗藥物的靶點、連接體技術、細胞毒性藥物的公開與否由投資者、合作者決定,因而無法掌握臨床試驗藥物的全部細節;原因二是與臨床試驗現狀不一定相符,藥物效力、患者招募、投資決策等風險因素時刻影響臨床試驗階段的變化,企業不一定及時將產品管線臨床研究的信息更新。從目前整理的信息來看,共有32種auristatins抗體-藥物偶合物經藥品監督管理機構審批進入臨床/臨床前試驗階段,其中:2種auristatins抗體-藥物偶合物進入臨床三期階段;11種auristatins抗體-藥物偶合物進入臨床二期階段;19種auristatins抗體-藥物偶合物進入臨床一期階段。
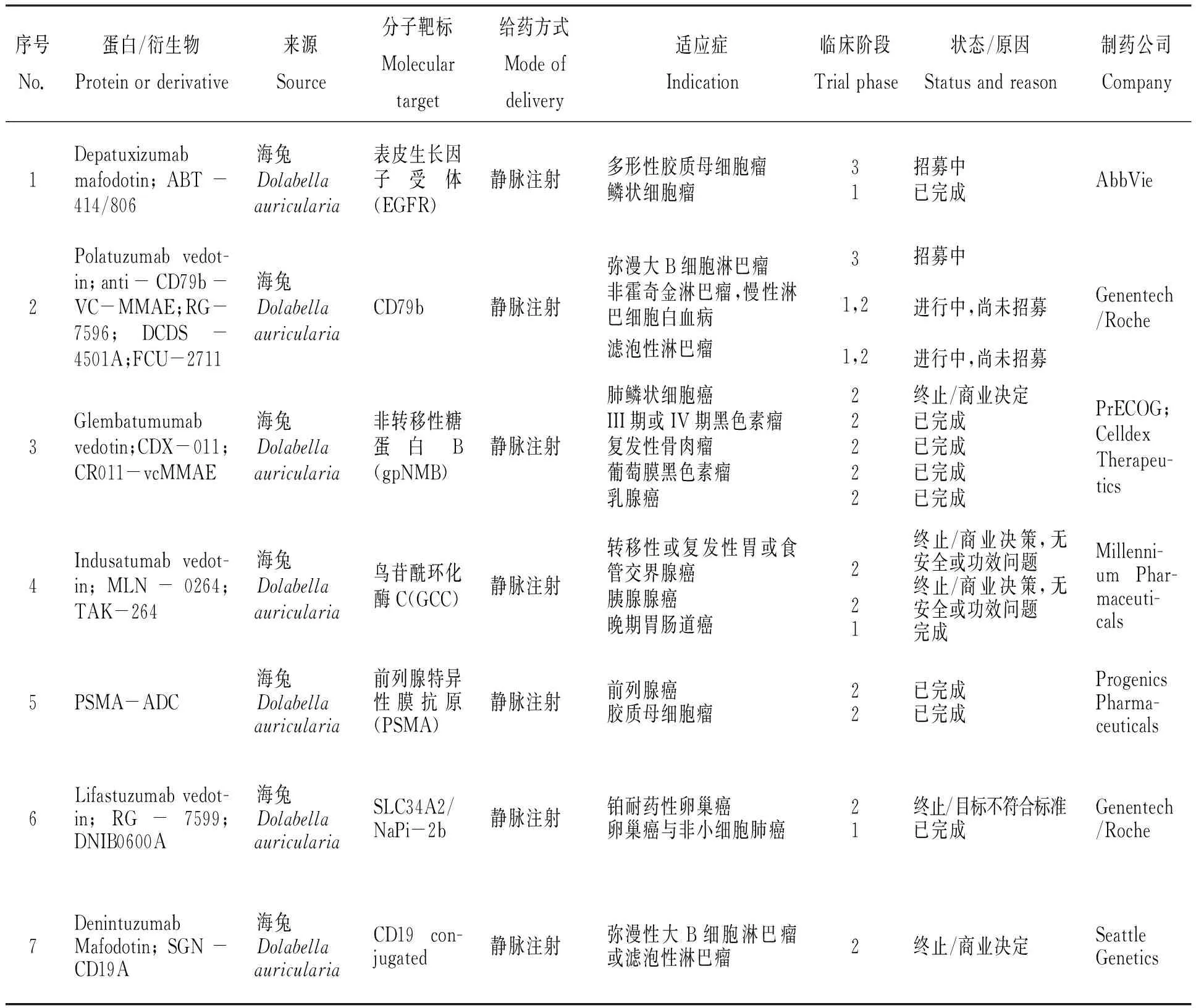
表3 經藥品監督管理機構審批進入臨床研究的auristatins抗體-藥物偶合物[12-37]
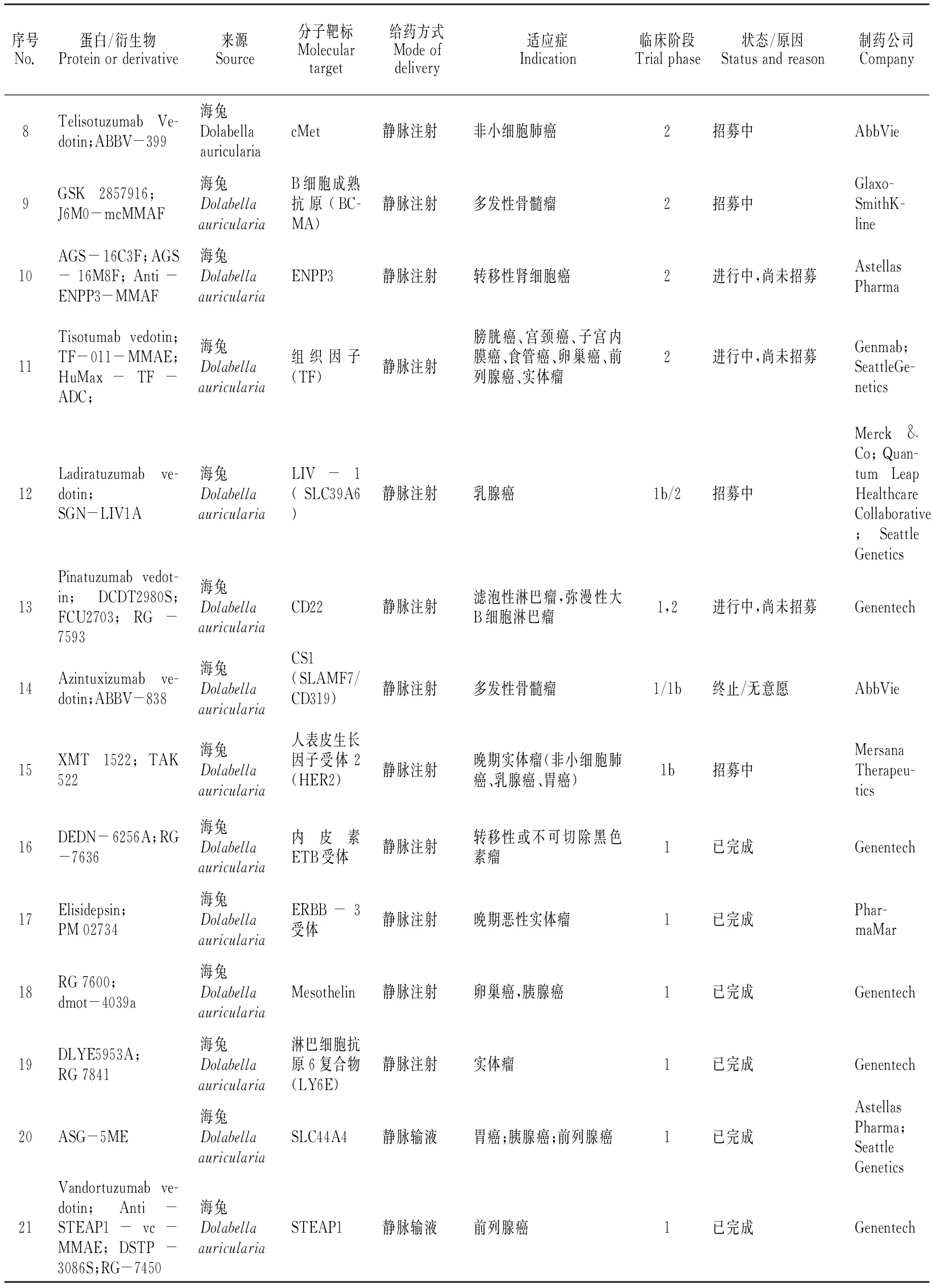
續表3
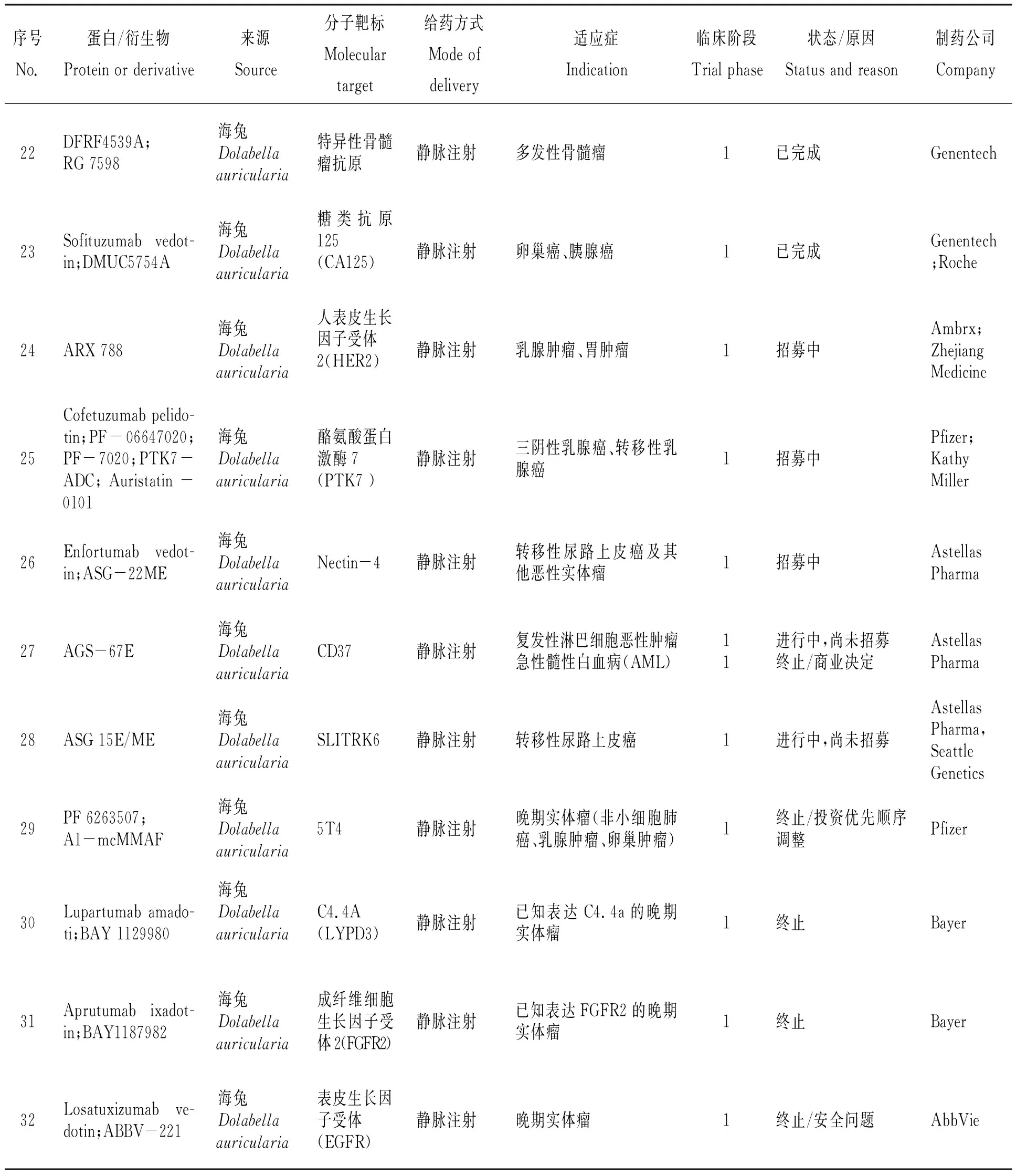
續表3
2 芋螺毒素及其衍生物
Cruz課題組于1985年首次報道了食魚性地紋芋螺(Conusgeographus)毒液多肽conotoxins(CTxs)的分離、鑒定[38]。Conotoxins是一類富含二硫鍵的神經肽(10 - 30個氨基酸殘基),根據與不同脊椎動物神經遞質/激肽受體和離子通道的特異性靶向親和能力,其分類為α-、δ-、κ-、μ-、ω-[39]。引人注意的是conotoxins通過翻譯后修飾(PTM),如C-端酰胺化、N-端焦谷氨酸化和關鍵氨基酸殘基的L到D異構化,以及廣泛的半胱氨酸交聯支撐,天然形成穩定的分子結構以對抗極端環境和蛋白酶水解。穩定、效力和靶向性等特質吸引了制藥企業和資本推動conotoxins從研發向臨床應用轉化。
2.1 經批準上市的芋螺毒素Ziconitide(Prialt?)
Ziconitide(又稱SNX-111)是由美國Elan公司研發的太平洋幻芋螺毒液多肽ω-MVIIA 的化學合成體,其與中樞神經系統N型電壓敏感性鈣離子通道高度親和,無需第二信使或G蛋白,故不會引起藥物依賴或成癮。經歷一系列臨床研究,美國食品藥品管理局(FDA)于2004年批準了Ziconitide以商品名Prialt?(圖1-B)上市,用于治療慢性疼痛(表2)[10]。盡管Prialt?具有3個二硫鍵穩定結構,但由于中樞神經受體的特殊性,其采用了鞘內注射的藥物輸送方式以增強中樞神經系統靶向效力。然而與腸外或口服給藥相比,鞘內注射具有內在風險[40],這使得Prialt?在患者接受度和市場滲透率方面受到了極大的限制。與同功效藥物對比,輝瑞公司口服活性鎮痛/抗癲癇藥物Neurontin?在2003年達到27億美元的銷售高峰[41],而Prialt?在2009財政年度的最終用戶銷售額僅為2 000萬美元,Azur Pharma在2010年僅以1 460萬美元收購了Prialt?歐洲版權[1],Prialt?在營銷方面不盡理想。
2.2 經批準進入臨床/臨床前試驗的芋螺毒素
由于conotoxins種類豐富,在中樞、外周神經系統中靶向位點眾多,且不易被蛋白酶水解,天然結構具有較強的藥物開發潛質,制藥界在臨床/臨床前研究中對其日益關注。表4總結了目前經審批進入臨床/臨床前試驗的芋螺毒素,靶向位點包括去甲腎上腺素轉運蛋白、GABAB a和 nAChR、神經降壓素受體、NMDA受體、CaV2.2通道、Shaker類型KV 通道,適用領域包括術后疼痛、神經病理性疼痛、難治性癲癇、心肌梗塞[1,35-37,42-45]。然而由于藥物缺乏功效、投資決策變更或撤資等因素影響,芋螺毒素均沒有進行超過臨床二期的研究。由此可見,與其他類型藥物相比,目前海洋動物蛋白藥物在中樞神經系統靶向開發成功率較低。
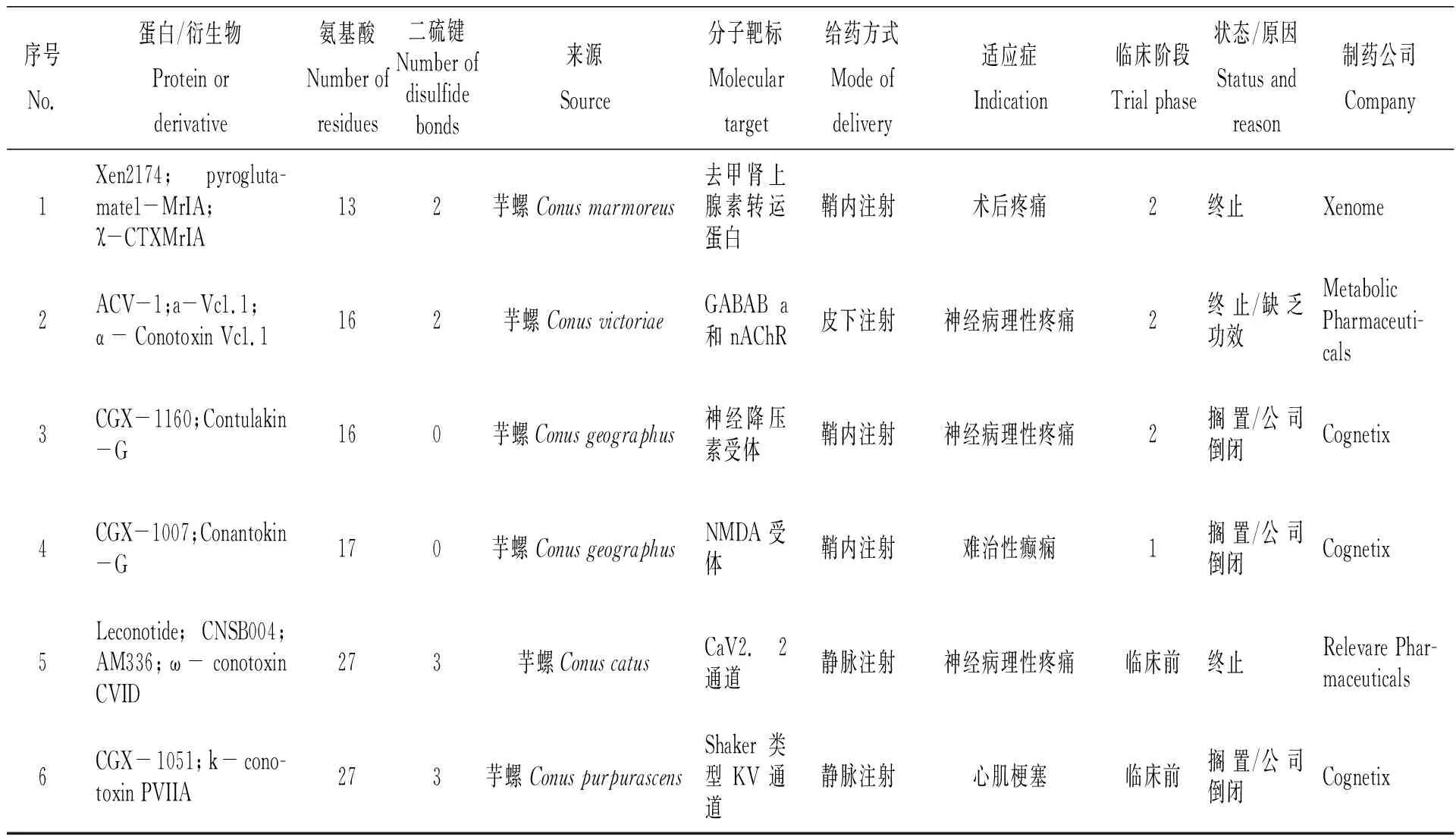
表4 經藥品監督管理機構審批進入臨床/臨床前研究的芋螺毒素[1,35-37,42-45]
3 海鞘毒素及其衍生物
Rinehart課題組于1981年報道了加勒比海海鞘(Trididemnumsolidum)提取物didemnins的分離、鑒定[46]。Didemnins是一類環形縮肽,具有較高的抗腫瘤活性,其中didemnin B的功能活性最強,通過特異性結合真核翻譯延長因子1A(eEF1A),啟動細胞凋亡,致使腫瘤細胞程序性死亡,成為美國第一個進入臨床試驗的海洋抗腫瘤天然產物[47]。Didemnin B(表5)已完成了針對復發性或難治性間變性星形細胞瘤或多形性膠質母細胞瘤的二期臨床試驗。不幸的是,該化合物由于患者中高發生率的過敏反應而顯示出高毒性,試驗已終止[48]。Didemnin同系物脫氫 didemnin B(又稱Plitidepsin)(表2、5)目前已完成了針對復發/難治性多發性骨髓瘤的臨床三期試驗[49],并向醫藥監管機構申請了上市許可。澳大利亞藥品管理局(TGA)于2018年12月批準了plitidepsin以商品名Aplidin?(圖1-C)上市,其將聯合地塞米松用于對其他療法治療失敗或有抵抗的復發性或難治性多發性骨髓瘤患者的治療[11]。
4 海綿活性多肽
Kashman課題組于1994年報道了南非海綿(Hemiasterellaminor)提取物中細胞毒性多肽hemiasterlin的分離、鑒定[50]。Hemiasterlin是一種三肽化合物,其通過與微管蛋白結合,使微管解聚,影響紡錘體形成,誘導有絲分裂停滯,促進細胞凋亡。為了提高作用效力,課題組合成了一系列衍生物,其中以Taltobulin(又稱HTI-286)和E 7974細胞毒性比天然產物hemiasterlin更佳,繼而進入臨床試驗階段。然而HTI-286因商業決策改變,不在制藥企業管線中體現而終止開發,E 7974則完成臨床一期試驗(表5)[9]。
5 海葵活性多肽
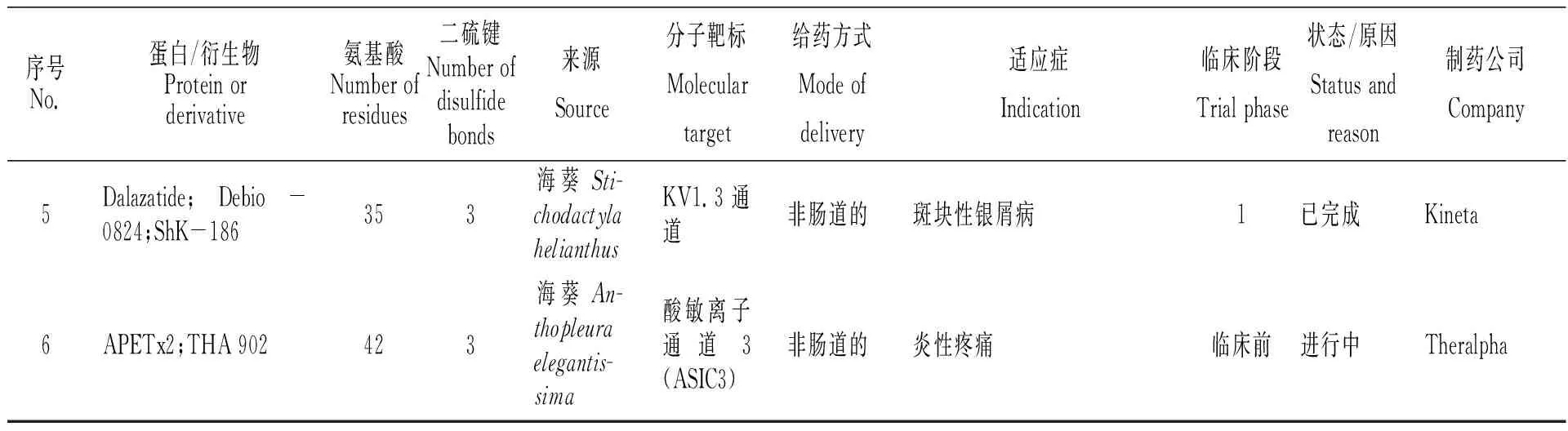
Karlsson課題組于1995年報道了加勒比海葵(Stichodactylahelianthus)毒液神經肽Stichodactyla toxin(ShK)的分離、鑒定[51]。ShK由35個氨基酸殘基組成,特異性作用于電壓門控Kv1.3鉀離子通道。2015—2017年,ShK類似物ShK-186(又稱Dalazatide)成為美國第一個進入臨床試驗的自身免疫疾病Kv1.3阻斷制劑,目前已完成臨床一期試驗(表5)[35-37]。Lazdunski課題組于2004年報道了海葵(Anthopleuraelegantissima)毒液神經肽APETx2的分離、鑒定[52]。APETx2是42個氨基酸、包含3個二硫鍵的神經多肽,特異性抑制感覺神經元中的酸敏感通道ASIC3。目前已進入臨床前試驗,適用于治療炎性疼痛(表5)[53]。
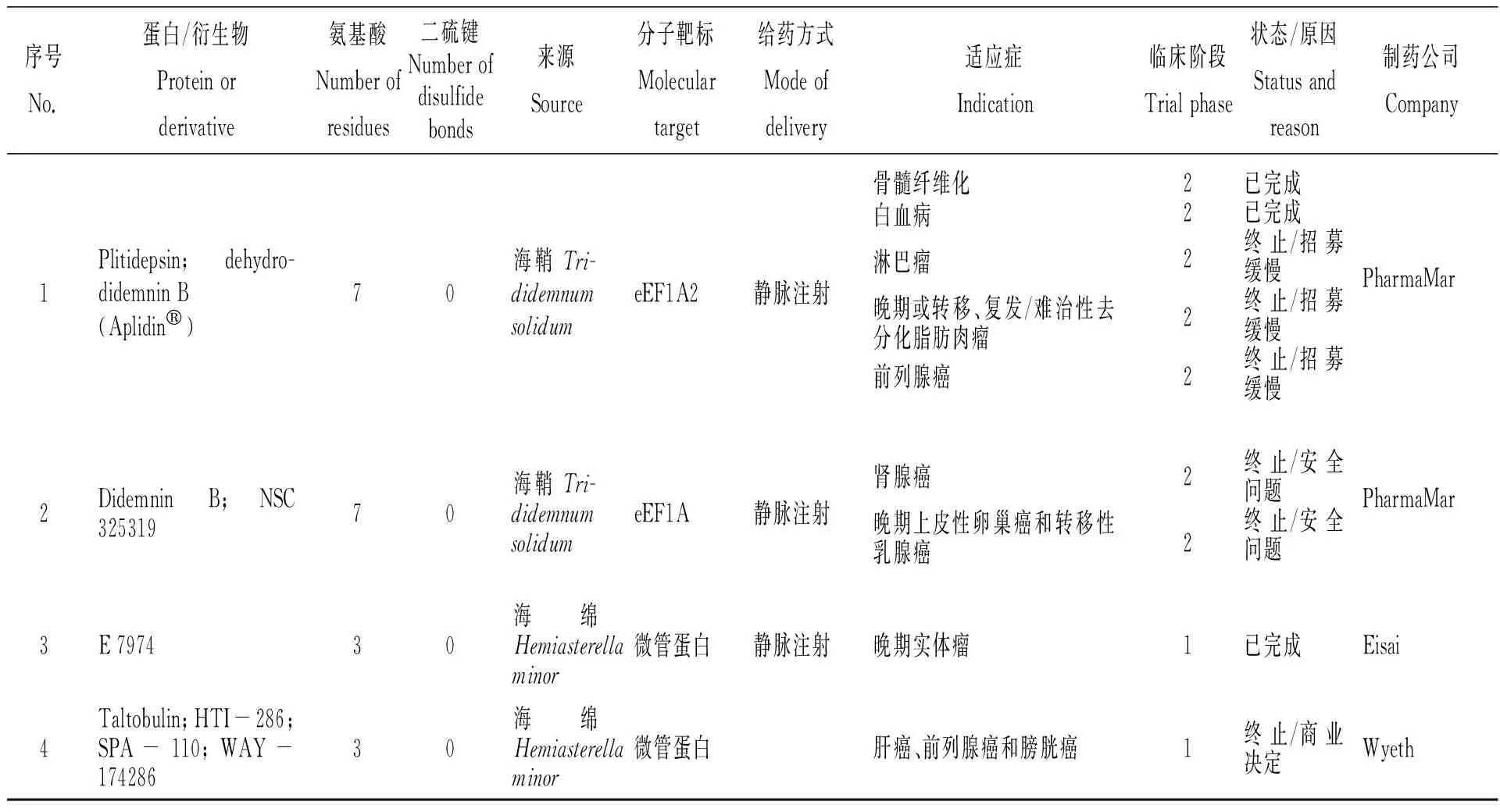
表5 經藥品監督管理機構審批進入臨床/臨床前研究的海鞘毒素、海綿活性多肽、海葵活性多肽[9,35~37,48-49,53]
續表5
6 結語與展望
本文對海洋動物來源蛋白藥物的最新研究進展進行了梳理。從功效領域來看,海洋動物蛋白主要歸類為細胞毒性蛋白、神經毒素蛋白和離子通道抑制劑。從藥物開發領域來看,海洋動物細胞毒性蛋白主要應用于抗癌和免疫藥物開發,市場轉化率好且呈現蓬勃發展的態勢;海洋動物神經毒素蛋白和離子通道抑制劑主要應用于治療慢性疼痛藥物開發,由于藥物代謝進入中樞神經系統的成功率僅為1%,故海洋動物神經毒素蛋白和離子通道抑制劑針對中樞神經系統靶標開發的衍生藥物成功率較低。從市場應用進展來看,目前共有3種海洋動物蛋白藥物(商品名Prialt?、Adcetris?和Aplidin?)獲得藥品監督管理機構批準成功上市,分別用于治療慢性疼痛、霍奇金淋巴瘤、系統性間變大細胞淋巴瘤和復發性或難治性多發性骨髓瘤。從藥物臨床進展最高階段來看,目前共有32種auristatins抗體-藥物偶合物、6種芋螺毒素、2種海鞘毒素、2種海綿活性多肽、2種海葵活性多肽經藥品監督管理機構審批進入臨床/臨床前試驗階段。其中2種auristatins抗體-藥物偶合物進入臨床三期階段,具有較好的藥品商業轉化預期和前景;11種auristatins抗體-藥物偶合物、3種芋螺毒素、2種海鞘毒素進入臨床二期階段;19種auristatins抗體-藥物偶合物、1種芋螺毒素、2種海綿活性多肽、1種海葵活性多肽進入臨床一期階段;2種芋螺毒素、1種海葵活性多肽經藥品監督管理機構審批進入臨床前試驗階段。
縱觀天然產物的藥物發展歷程,從20世紀70、80年代美國癌癥研究中心(NCI)和美國農業部(USDA)啟動轟轟烈烈的陸源、海源天然藥物篩選項目,到90年代制藥企業紛紛終止天然產物項目,再到本世紀初高通量篩選和組合化學以及近十年來發展迅猛的細胞毒性藥物-單克隆抗體聯合藥物,海洋動物蛋白藥物完整經歷了技術更新、資金和時間的考驗,在惡性腫瘤、慢性疼痛等疾病治療以及神經系統離子通道特異性診斷方面取得了實實在在的成果。據此,相信在未來,隨著質譜、核磁共振波譜、轉錄組學技術的發展,將有越來越多海洋動物來源的活性蛋白被發現;隨著單克隆腫瘤抗體靶向技術和突破血腦屏障緩釋技術水平的提升,海洋動物蛋白藥物的效能與給藥方式的瓶頸將得以突破。我們期待更多的藥物從自然海洋走向醫護救治。
