荔枝與龍眼中噻蟲啉、螺蟲乙酯及代謝物的高效液相色譜-串聯質譜分析
2019-03-08 02:38:06王思威曾廣豐劉艷萍王瀟楠孫海濱
分析測試學報 2019年2期
王思威,曾廣豐,劉艷萍,王瀟楠,孫海濱*
(1.廣東省農業科學院植物保護研究所 廣東省植物保護新技術重點實驗室,廣東 廣州 510640;2.廣東檢驗檢疫技術中心,廣東 廣州 510623)
荔枝(LitchichinensisSonn.)和龍眼(DimocarpuslonganLour.)均為無患子科植物,屬亞熱帶地區重要的常綠果樹,主要產于廣東、廣西、福建等地[1-4]。荔枝含有豐富的維生素C、維生素E、多糖、類黃酮等成分;龍眼具有補血益氣、安神養心之功效,含有糖類、維生素、酚類、多糖等成分[1-4]。南方高溫的氣候特點使得荔枝、龍眼樹的蟲害嚴重,而煙堿類殺蟲劑噻蟲啉和季酮酸類殺蟲劑螺蟲乙酯因具有較強的內吸作用,對荔枝、龍眼樹的薊馬、螨類等刺吸式口器害蟲具有良好防效。其中,螺蟲乙酯因具有雙向內吸傳導的獨特性能,使其可在植物體內移動,抵達葉面和樹皮,有效阻止害蟲的卵和幼蟲的生長發育,增強防治效果。螺蟲乙酯在植物體內的降解產物主要有BYI08330-烯醇糖苷(B-glu)、BYI08330-醇酮(B-keto)、BYI08330-烯醇(B-enol)、BYI08330-羥基(B-mono)4種。噻蟲啉對蜜蜂群落有顯著影響(荔枝、龍眼主要靠蜜蜂傳粉),且可增加肝臟腫瘤發病率[5];螺蟲乙酯及代謝物可引起動物和人的眼部刺激和潛在皮膚過敏效應,且對地下水的潛在影響未知[6]。通過建立荔枝和龍眼中螺蟲乙酯和噻蟲啉的分析方法,可為后續對荔枝和龍眼中2種殺蟲劑的膳食攝入風險評估及對蜜蜂的風險評價提供研究基礎。當前噻蟲啉和螺蟲乙酯及代謝物殘留測定的研究主要集中于番茄、菠菜、柑橘等,暫未發現荔枝和龍眼中的相關報道。
目前,關于噻蟲啉的檢測方法主要有液相色譜法(HPLC)、液相色譜-質譜法(LC-MS/MS)[7-11],前處理方法主要有液液分配、QuEChERS凈化、弗羅里硅土填充柱或固相萃取柱凈化,定量下限為0.01~0.05 mg/kg[12-15]。螺蟲乙酯的檢測方法有HPLC法、氣相色譜-質譜法(GC-MS)和LC-MS/MS法,其代謝物均采用液質聯用分析法;前處理方法包括液液分配萃取、固相萃取(SPE)、分子印跡和QuEChERS凈化等[16-22],定量下限為0.005~0.05 mg/kg[23-26]。但液液分配萃取、填充柱凈化等前處理方法的有機溶劑用量大,且操作繁瑣,費時費力;固相萃取和分子印跡技術的成本較高。QuEChERS法的獨特優勢(快速、簡便、通用性強)使其對水果、蔬菜等基質中的凈化效果十分突出[27-29]。本研究以荔枝和龍眼為研究對象,建立了噻蟲啉、螺蟲乙酯及代謝物的QuEChERS/高效液相色譜-串聯質譜分析方法,為不同水果中噻蟲啉、螺蟲乙酯及其代謝物殘留的同時檢測提供了簡便、快速、精準的新方法。
1 實驗部分
1.1 儀器與試劑
AB SCIEX 4000Q 三重四極桿質譜儀,配電噴霧離子源(ESI)(美國AB SCIEX公司);Agilent LC-1200高效液相色譜儀(美國安捷倫公司);Milli-Q超純水機(美國Millipore公司);GTR22-1離心機(北京時代北利離心機有限公司);OA-SYS氮吹儀(美國Organomation Associates公司);XW-80A渦旋儀(上海精科有限公司)。
乙腈(色譜純,美國Fisher公司);甲酸(色譜純,美國Fluka公司);無水硫酸鎂、氯化鈉(分析純,國藥集團化學試劑有限公司);十八烷基鍵合硅膠(C18)、N-丙基乙二胺(PSA)、石墨化碳黑(GCB)吸附劑(上海安譜實驗科技股份有限公司)。標準品:噻蟲啉(純度99.0%,美國Chem Service公司);螺蟲乙酯及代謝物B-glu、B-keto、B-enol、B-mono的純度分別為99.2%、89.6%、92.8%、99.8% 和98.2%(德國Ehrenstorfer GmbH)。
1.2 標準溶液的配制
標準儲備溶液:分別準確稱取適量噻蟲啉、螺蟲乙酯及4種代謝物標準品,用乙腈配制成1 000 mg/L的標準儲備溶液,于4 ℃避光保存,待用。
混合標準溶液:分別準確吸取1 mL噻蟲啉、螺蟲乙酯及4種代謝物的標準儲備溶液于同一10 mL容量瓶中,用乙腈定容至10 mL,配制成100 mg/L的混合標準溶液。
1.3 樣品前處理
準確稱取荔枝、龍眼樣品5.0 g(精確至0.01 g),置于50 mL離心管中,加入10 mL乙腈進行提取,以10 000 r/min 勻質1 min后,加入2 g無水硫酸鎂和1 g氯化鈉,劇烈振蕩,渦旋1 min,再以5 000 r/min離心2 min。取2 mL乙腈層溶液,置于裝有0.3 g無水硫酸鎂、0.025 g C18和0.025 g GCB的離心管中,劇烈振蕩,渦旋10 s,再以10 000 r/min離心2 min。取上清液過0.22 μm有機濾膜,待測定。
1.4 色譜-質譜條件
色譜條件:Poroshell-120 EC-C18柱(150 mm×3.0 mm,2.7 μm);柱溫為35 ℃;流動相為0.1%(體積分數)甲酸水溶液-乙腈(體積比25∶75),等度洗脫5 min;流速為0.4 mL/min;進樣量為5 μL。
質譜條件:離子源為ESI+;掃描模式為多反應監測模式(MRM);離子源溫度為550 ℃;氣簾氣壓力為0.2 MPa;毛細管電壓為5 500 V;霧化氣壓力為0.3 MPa;加熱輔助氣壓力為0.3 MPa。其他質譜參數見表1。
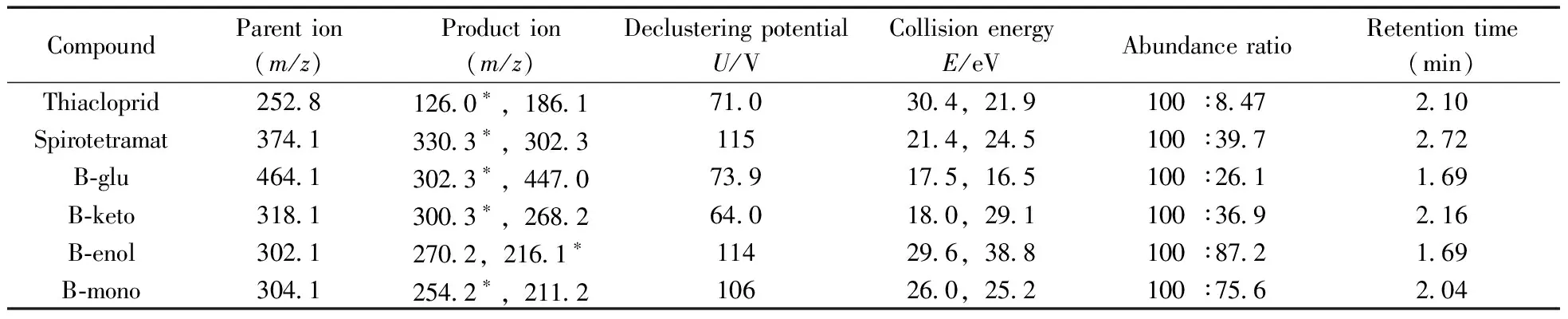
表1 噻蟲啉、螺蟲乙酯及代謝物的質譜參數與保留時間Table 1 Mass spectra parameters and retention times of thiacloprid,spirotetramat and its four metabolites
*quantitative ion
2 結果與討論
2.1 前處理條件的優化
2.1.1提取溶劑的選擇考察了乙酸乙酯、乙腈和乙腈-乙酸乙酯(體積比1∶1)作為提取溶劑時對6種目標化合物的提取效率。結果顯示,以乙腈為提取溶劑時,6種目標農藥的提取率為80.7%~103%;以乙酸乙酯為提取溶劑時,B-enol的回收率僅為13%;而乙腈-乙酸乙酯(1∶1)為提取溶劑時,B-enol的回收率為3.3%。這可能是由于乙酸乙酯溶于水,導致其不能從基質中完全提取極性較強的B-enol。因此,最終選擇乙腈作為提取溶劑。
2.1.2吸附劑的選擇實驗考察了QuEChERS前處理方法的常用吸附劑(PSA、C18、GCB、PSA+C18、PSA+GCB、C18+GCB)對噻蟲啉、螺蟲乙酯及4種代謝物的吸附效果。結果顯示:PSA吸附劑對B-enol和B-glu的回收率較低(25.6%~40.8%);PSA+C18組合對B-enol和B-glu的回收率極低(15.0%~24.3%);PSA+GCB組合對B-glu的回收率較低(30.2%~34.4%);采用C18、GCB或C18+GCB組合對上述6種化合物的回收率均在80%以上。C18吸附劑是硅膠基質鍵合十八烷基填料,其比表面積大,吸附能力較強,能夠去除脂肪、脂類等非極性干擾物;GCB可顯著去除樣品中的色素。由于荔枝和龍眼樣品中的色素、糖類等物質較多,采用單一吸附劑不能有效去除雜質,因此本研究選用0.025 g C18+0.025 g GCB吸附劑組合,可使噻蟲啉、螺蟲乙酯及4種代謝物獲得良好的吸附效果及回收率。
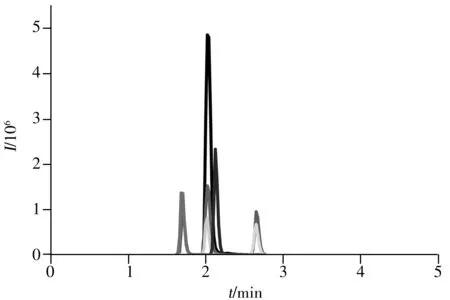
圖1 Poroshell-120 EC-C18色譜柱對噻蟲啉、螺蟲乙酯及代謝物的分離色譜圖
2.2 色譜柱的選擇
在乙腈-0.1%甲酸流動相體系下,比較了Poroshell-120 EC-C18(150 mm×3.0 mm,2.7 μm)、XBridge C18(100 mm×2.1 mm,3.5 μm)、Atlantis T3 C18(100 mm×3.0 mm,3.0 μm) 3種色譜柱對6種目標物的分離效果。結果顯示:Poroshell-120 EC-C18柱對目標物的分離效果略好,峰形對稱(圖1);XBridge C18柱對目標物的分離度較差;Atlantis T3 C18柱對目標物分離的峰形不對稱,產生了拖尾現象,影響定量結果。綜上,選擇Poroshell-120 EC-C18作為本研究的色譜分離柱。
2.3 流動相的選擇
考察了甲醇-水、甲醇-0.1%甲酸水溶液、乙腈-水、乙腈-0.1%甲酸水溶液4種流動相體系對目標化合物的分離度及響應強度。結果顯示:以甲醇為有機相時,B-glu的峰形不對稱、峰展寬,影響定量結果。而以乙腈為有機相時,在水相中加入0.1%甲酸可以促進[M+H]+離子峰形成,提高目標農藥檢測的靈敏度。因此,流動相選用乙腈-0.1%甲酸水溶液。
2.4 質譜條件的優化
考察了正負離子掃描模式對6種目標化合物的離子化效果,發現目標物在正離子模式下響應更高,為提高目標化合物的離子化效率,對源內參數進行了優化。在正離子模式下,采用針泵進樣的方式對化合物進行全掃描,通過一級質譜掃描獲取穩定的[M+H]+分子離子,確定母離子后,在MRM模式下對化合物的去簇電壓(DP)進行優化;在二級質譜中,母離子發生斷裂或重排等裂解反應,產生不同的m/z離子碎片,選擇響應最高且干擾最少的2個碎片離子為定性和定量離子,分別優化其碰撞能量(CE)。優化后的質譜條件如“1.4”所示,二級質譜圖見圖2。

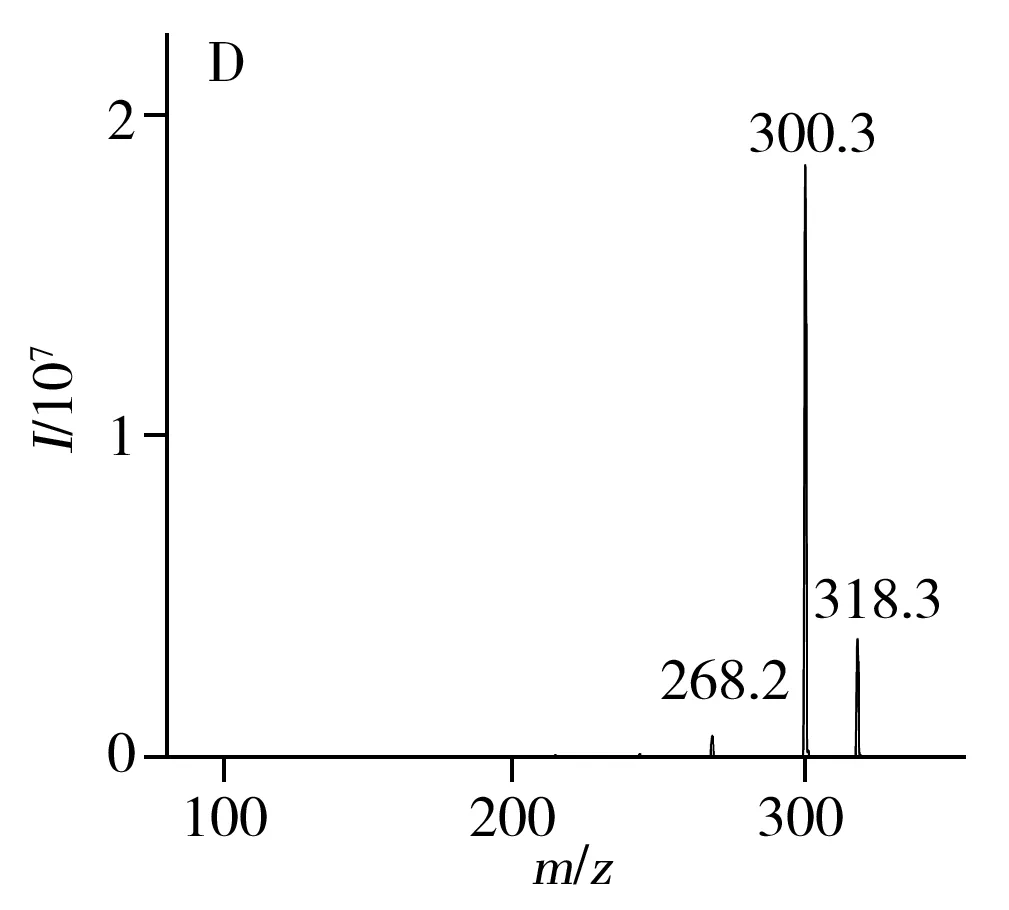
2.5 基質效應
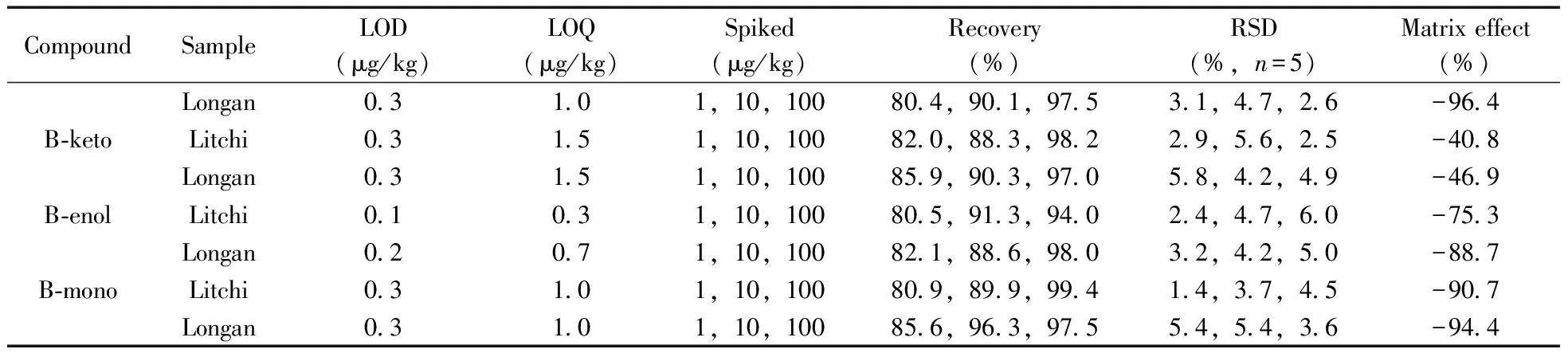
采用HPLC-MS/MS分析樣品時,基質效應(ME)是影響定量結果準確性的重要因素。本實驗采用下式計算基質效應:ME(%)=[(mmatrix/msolvent)- 1)]× 100%,其中mmatrix為基質匹配標準曲線的斜率,msolvent為純溶劑標準曲線的斜率。當ME為正值時,表示存在基質增強效應;ME為負值時,表示存在基質抑制效應;ME=0時,表示不存在基質效應。實驗結果表明(表2),噻蟲啉、螺蟲乙酯及其4種代謝物在荔枝和龍眼中的ME均為負值,表現為基質抑制作用,其中B-keto在荔枝和龍眼中的基質效應絕對值低于47%,其它化合物的基質效應絕對值為75.3%~96.7%,表明B-keto的基質抑制效應相對較強,其它化合物則相對較弱。本實驗采用基質匹配標準溶液校正基質效應,從而確保實驗結果的準確性和可靠性。
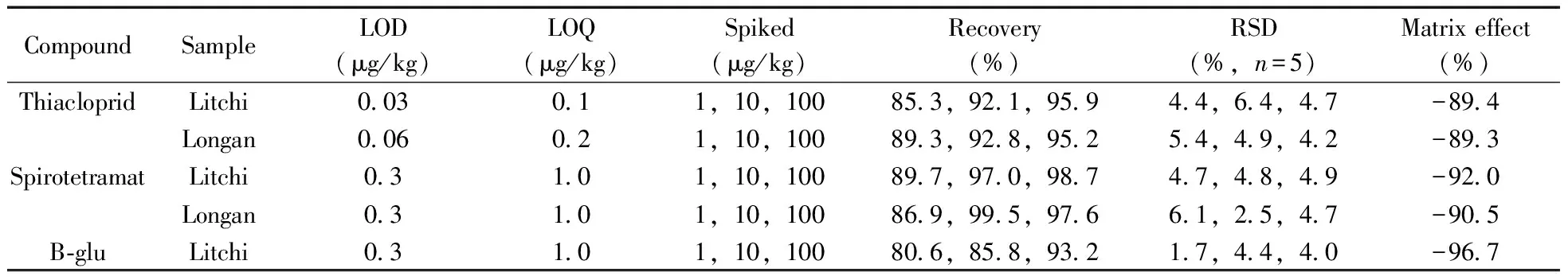
表2 噻蟲啉、螺蟲乙酯及代謝物的檢出限、定量下限、回收率、相對標準偏差及基質效應Table 2 LODs,LOQs,recoveries,RSDs and matrix effects of thiacloprid,spirotetramat and its four metabolites in litchi and longan samples
(續表2)
CompoundSampleLOD(μg/kg)LOQ(μg/kg)Spiked(μg/kg)Recovery(%)RSD(%,n=5)Matrix effect(%)Longan0.31.01,10,10080.4,90.1,97.53.1,4.7,2.6-96.4B-ketoLitchi0.31.51,10,10082.0,88.3,98.22.9,5.6,2.5-40.8Longan0.31.51,10,10085.9,90.3,97.05.8,4.2,4.9-46.9B-enolLitchi0.10.31,10,10080.5,91.3,94.02.4,4.7,6.0-75.3Longan0.20.71,10,10082.1,88.6,98.03.2,4.2,5.0-88.7B-monoLitchi0.31.01,10,10080.9,89.9,99.41.4,3.7,4.5-90.7Longan0.31.01,10,10085.6,96.3,97.55.4,5.4,3.6-94.4
2.6 方法學評價
2.6.1線性范圍、檢出限與定量下限在1~500 μg/L范圍內,以基質匹配標準溶液的質量濃度(x,μg/L)為橫坐標,對應的響應值(y)為縱坐標作圖,經最小二乘法得噻蟲啉、螺蟲乙酯及4種代謝物的線性方程。結果顯示,6種目標物的線性關系良好,相關系數(r2)均大于0.990。分別以3倍和10倍信噪比(S/N)確定方法的檢出限(LOD)和定量下限(LOQ)。由表2可知,本方法對噻蟲啉的LOD和LOQ分別為0.03~0.06 μg/kg和0.1~0.2 μg/kg,均低于煙草[9](0.5 μg/kg和0.16 μg/kg)和花生[14](1~5 μg/kg)中的報道值;對螺蟲乙酯及其4種代謝物的LOD和LOQ分別為0.1~0.3 μg/kg和0.3~1.5 μg/kg,均低于動物源食品[21](10~50 μg/kg,30~300 μg/kg)、牛奶[12](0.125~0.75 μg/kg,0.425~2.5 μg/kg)和番茄[15](LOD為0.03~1.6 μg/kg)等基質中的報道值。
2.6.2回收率與相對標準偏差在荔枝和龍眼空白樣品中添加1、10、100 μg/kg 3個濃度水平的6種目標物混合標準溶液,每個加標水平重復5次,并作空白對照。6種目標物在荔枝和龍眼中的平均加標回收率為80.4%~99.5%,相對標準偏差(RSD)為1.4%~6.4%(表2)。荔枝空白樣品加標的色譜圖見圖3。本方法具有較高的回收率和良好的精密度,可滿足荔枝和龍眼樣品中噻蟲啉、螺蟲乙酯及4種代謝物的檢測要求。
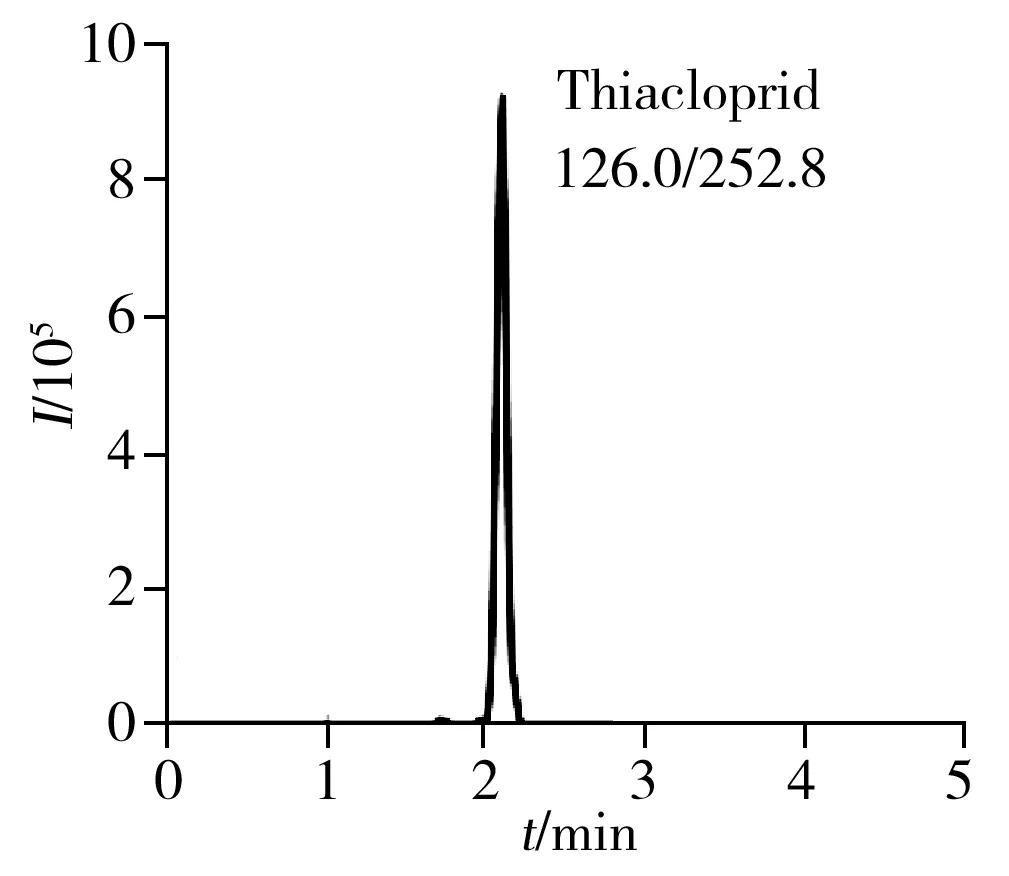
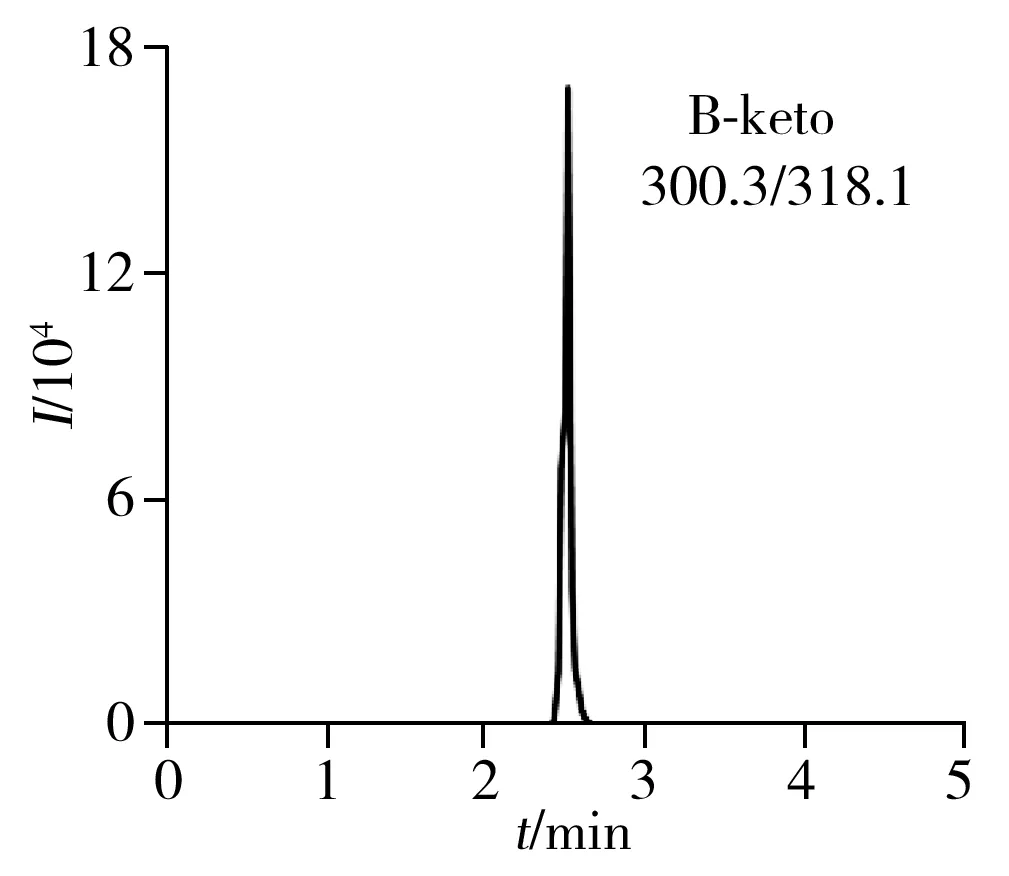
2.7 方法的應用
采用本方法對市場中隨機購買的20份荔枝和龍眼樣品進行檢測,均未檢出噻蟲啉、螺蟲乙酯及4種代謝物的殘留。
3 結 論
本文在對樣品前處理和儀器分析條件優化的基礎上,建立了測定荔枝和龍眼中噻蟲啉、螺蟲乙酯及4種代謝物殘留的HPLC-MS/MS方法。應用優化的QuEChERS方法進行樣品前處理,操作簡便、可靠,方法檢出限為0.03~0.3 μg/kg,定量下限為0.1~1.5 μg/kg。荔枝和龍眼中噻蟲啉、螺蟲乙酯及4種代謝物在1、10、100 μg/kg水平下的平均加標回收率為80.4%~99.5%,RSD為1.4%~6.4%。本方法可用于荔枝和龍眼等水果中噻蟲啉、螺蟲乙酯及代謝物的定性定量分析。
