基于表面氧空位的光催化固氮材料
2019-03-20 06:23:42毛成梁張禮知
中國材料進展 2019年2期
毛成梁,張禮知
(華中師范大學化學學院 環境與應用化學研究所 農藥與化學生物學教育部重點實驗室,湖北 武漢430079)
1 前 言
氮是生命體不可或缺的元素,雖然地球大氣中含有大量分子氮——氮氣(N2, 78%),但是由于N—N鍵鍵能高(940 kJ/mol)、質子親和力差和離子化能高,分子氮難以解離而不能直接被生命體利用。因而,工業Harbor-Bosch過程需要在高溫高壓(15~25 MPa, 300~550 ℃)、Fe或釕(Ru)催化劑存在下才能克服N2解離活化高能壘,實現原子態N和H組裝,高效合成氨(NH3)[1-6]。但是該過程每年消耗1%~2%地球總能量,排放約2.3 t CO2,對全球氣候變化和能源影響巨大。因而,探索合成氨新途徑意義重大。
自然界生物固氮也是一種可大量固氮的方式,雖然該過程與工業合成氨同樣是高耗能過程(固定1 mol N2約消耗500 kJ能量[7]),但依靠固氮酶FeMo輔因子吸附分子氮并傳遞電子使N—N鍵伸長而被質子化,在逐步還原質子化中伴隨N—N鍵鍵級降低和N—N鍵斷裂,最終實現溫和條件生物固氮[8]。生物固氮讓我們認識到催化劑具有N2吸附位點,并存在電子供體還原分子氮,就可能實現溫和條件固氮。
基于生物固氮過程的認識,早期研究模仿固氮酶作用機制設計了一系列固氮新途徑。其中,具有光合作用特性的異相光催化固氮被認為是極具潛力的“綠色”新途徑。這是因為光催化策略使用廉價易得的半導體材料為催化劑,以N2和水為底物,在可持續能源太陽光的驅動下即可生成氨和氧氣,該過程不需要昂貴的還原劑、有機溶劑和額外的化石能源和電能供給[9]。然而,光催化固氮受限于其較低的固氮效率,這是因為半導體對光能吸收利用有限、并且被吸收的光子也很難有效轉化為能參與反應的光生載流子——大部分載流子在催化劑體相復合而不能參與表面固氮反應[10-12]。此外,與常見的光催化產氫和CO2還原反應相比,光催化固氮更具挑戰,這是因為N2還原質子化的中間產物能量極高。例如,理論上N2還原、還原質子化都需要極負的電位,遠超出常見半導體(如TiO2、ZnO、CdS)導帶所能達到的還原電位[13]。如何提升材料光能利用效率,預活化分子氮降低其還原質子化能壘、提高效率是光催化固氮亟待解決的問題。
針對以上問題,作者團隊提出在BiOBr光催化劑表面構造活性位點氧空位,提升光催化固氮活性,并闡明了氧空位介導的分子氮活化機制[14]。接下來,作者耦合半導體基光催化材料和熱合成氨活性中心Ru,構建氧空位促進的Ru基合成氨光熱催化劑,極大地提高光催化固氮效率,在常壓下達到與傳統熱合成氨相當的效率,使光催化固氮成為一種有潛力的高效合成氨新途徑[15]。本文系統總結了近年來圍繞光催化固氮材料表面缺陷活性位點構建的研究成果,揭示了表面活性位點尤其是缺陷位誘導的分子氮活化原理,以及在表面缺陷位基礎上構建協同活性中心,如過渡金屬納米晶和單原子,促進分子氮活化,提高光催化固氮效率。借此加深理解氧空位活性位點和分子氮活化之間的關聯,為設計新型高性能光催化固氮催化劑提供理論支持。
2 光催化固氮
光催化固氮最早報道于1977年,Schrauzer等[9]利用0.2 g金紅石TiO2為催化劑,N2氣氛下太陽光照射2 h水溶液中僅生成0.5 μmol NH3和0.09 μmol N2H4。這是因為半導體TiO2僅被能量大于其帶隙的光子激發,而金紅石TiO2帶隙為3.03 eV,僅能利用太陽能中不足5%的紫外光,因而效率低。通過2%(質量分數)Fe2O3摻雜可將TiO2固氮活性提高兩倍以上,其中Fe2O3摻雜TiO2不僅改性光催化劑能帶結構,優化光能吸收利用,其本身也具有光催化固氮活性,可輔助提升反應效率。采用金屬摻雜思路,Schrauzer等發現Fe,Mo,Co,Ni摻雜促進TiO2固氮活性,其促進效果依次遞減,而Pd,Pt,Au,V,Cr,Pb,Cu摻雜表現出抑制效果[9]。這些結果表明:① 除TiO2和Fe2O3以外,其他金屬元素化合物也能實現光催化固氮;② 增強光催化劑光吸收利用可提高固氮效率。
因而后續大量研究集中于探索光催化固氮新材料,全面總結見于Li和Kong等的綜述文章[16],這些材料主要包括Ti,Fe,Zn,Ga,Ta,Bi,C,Au,Os,Ru,Cr,Si,W,Mo,Cu,V等元素的單質、氧化物、鹵氧化物、硫化物、氫氧化物、氮化物和碳化物。此外,調控材料光吸收和光生載流子生成分離效率也是增強固氮性能的基本策略,包括能帶調控、多組分半導體復合、等離子共振金屬負載等,相關綜述見于Qiao等的綜述文章[17]。
隨著大量材料被發現具有光催化固氮活性,從反應熱力學角度選擇性設計催化材料成為固氮發展方向,主要包括選擇合適的金屬活性中心降低反應活化能,并耦合半導體能帶結構實現光生電子由光催化劑到表面含N物種的有效傳遞。以上催化劑設計主要依賴于光催化劑中金屬元素的本征特性,例如過渡金屬(TM)d軌道對N2的吸附活化能力,這些工作繼承了Harbor-Bosch合成氨的研究經驗,通過替換金屬元素實現反應活性的調控,相關固氮催化劑熱力學從頭設計綜述見于Hatzell和Medford的文章[18]。此外,從熱力學上直接越過固氮反應中的高能壘步驟如N2單電子還原也是可行的固氮策略。Hamers等[13]報道了H封端的寶石在深紫外照射下產生具有極強還原性的水合電子,可直接還原N2而固氮。但是這一策略要求催化劑導帶位置高于絕對真空能級(負電子親和性),一般半導體材料難以達到。
區別于以往光催化固氮綜述論文,本文主要針對作者團隊2015年首次報道并獲得廣泛關注的表面氧空位誘導光催化固氮性能增強策略(圖1),著重歸納不同材料中缺陷(空位)尤其是氧空位在光催化固氮中的一般作用機制。
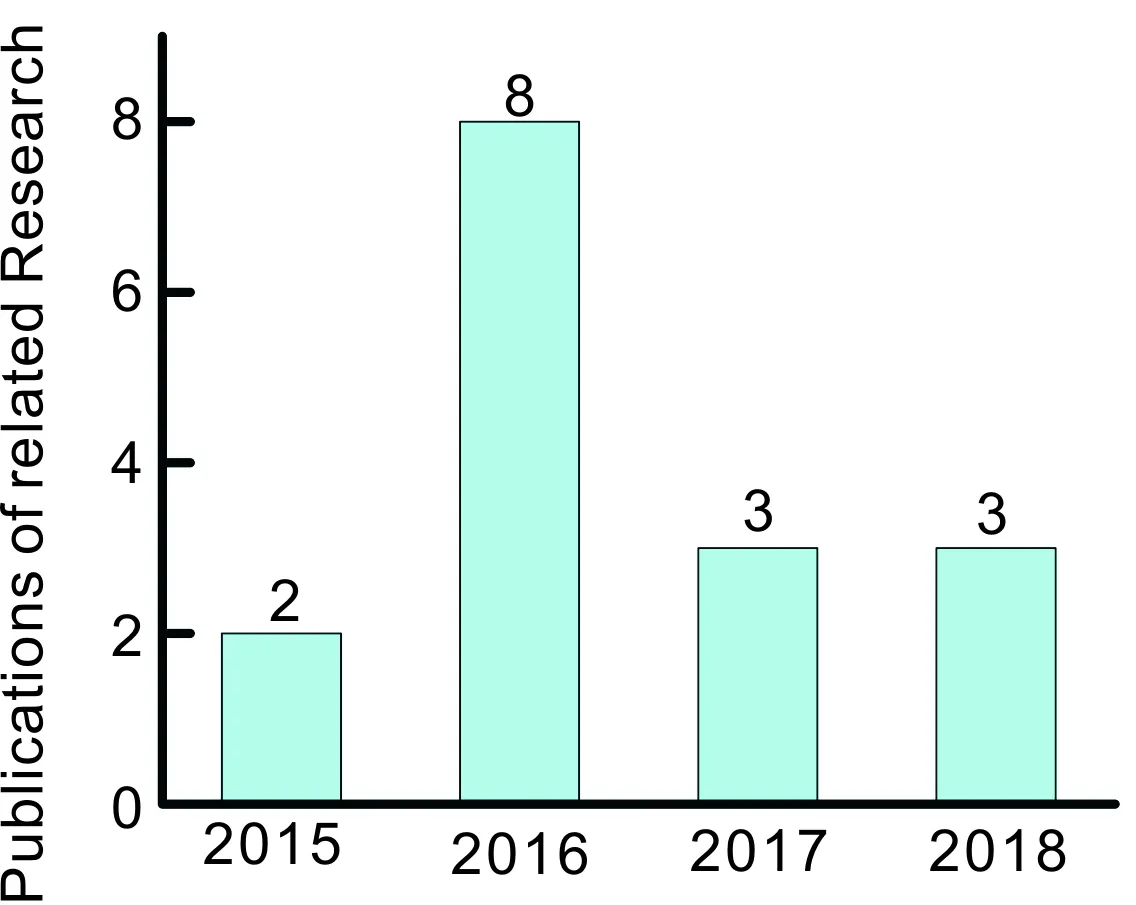
圖1 2015年以來每年發表的基于缺陷增強光催化固氮的研究論文數量Fig.1 The number of publications per year on vacancy-based photocatalytic nitrogen fixation enhancement since 2015
3 缺陷位促進的光催化固氮
隨著納米技術的發展,納米結構調控被發現能極大程度改變某一材料的催化性能,而其中最具吸引力的結構調控就是構建表面缺陷位,因為表面缺陷位通常作為小分子吸附活化位點而成為催化活性中心。作者團隊首次提出BiOBr表面氧空位作為固氮活性位點,吸附活化分子氮而降低N2還原質子化能壘,并在光照下光生電子轉移到表面氧空位而實現高效固氮。之后,氧空位或其他缺陷位對固氮活性增強機理在不同材料體系(包括Mo、Bi、Ti、W、C3N4等)得到驗證,因而構建氧空位(缺陷位)可能成為一種固氮活性增強通用策略。
3.1 氧空位的原子結構和電子結構
氧化物中晶格氧脫出位點被稱作氧空位(Olattice→OV+1/2 O2),它是一種最常見的表面陰離子缺陷,一般可通過惰性氣氛或還原氣氛中加熱、紫外光輻射等方式制備。表面氧空位不僅在幾何結構上構建原子級缺陷,而且暴露大量配位不飽和金屬原子而具有特殊電子結構。以BiOCl為例,由于馬德隆勢(Madelung potential[19])的存在,氧空位的剩余電子局域在氧空位周圍而不是均勻分布在整個晶體內部,通常選擇性局域在氧空位臨近Bi原子而形成氧空位-低價態Bi共存形式。在能帶結構上通常表現為導帶底下方出現由Bi的p軌道和O的p軌道組成的雜質能級,因而表面氧空位拓寬BiOCl的光響應范圍,同時抑制光生電子和空穴的復合。
氧空位結構高度依賴于其周圍原子幾何構型,因而氧空位結構調控可通過特定手段如暴露晶面和調控氧空位濃度實現。BiOCl{001}面和{010}面具有不同構型的暴露原子,因而對應晶面的氧空位具有不同原子構型,{001}面氧空位近鄰2個Bi原子和4個O原子,而{010}面氧空位近鄰3個Bi原子和2個Cl原子(圖2a和2b)[20]。而在同一晶面{001}面上,BiOCl氧空位濃度和氧空位周圍原子結構具有濃度-結構依賴關系,增加氧空位濃度,則使氧空位臨近Bi-Bi間距縮短,并使氧空位局域電子在氧空位臨近的2個Bi原子上更離域(圖2c)[21]。
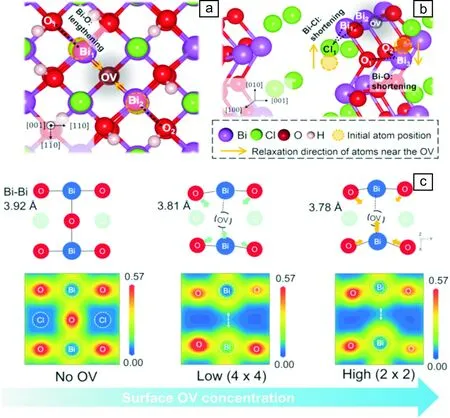
圖2 {001}晶面(a)和{010}晶面(b)BiOCl氧空位的原子構型[20];{001}晶面BiOCl氧空位濃度和原子、電子結構對應關系(c)[21] Fig.2 Atomic configuration of oxygen vacancy (OV) on the {001} (a) and {010} (b) facet of BiOCl[20]; OV atomic and electronic structure dependent on OV concentration of BiOCl {001}(c)[21]
3.2 氧空位對分子氮的吸附活化
由于N2的N—N鍵鍵能高,分子氮活化通常是反應速控步,如何促進分子氮活化是提高固氮效率的關鍵[1, 22]。活性中心如Fe、Ru、Mo帶負電則更容易實現N2吸附活化,這是因為它們的d軌道與N2的p軌道形成σ鍵和π鍵,其中由過渡金屬(TM)d軌道向N2的p軌道的π鍵電子反饋作用(backdonating effect)能削弱N≡≡N三鍵同時增強TM—N鍵的強度[23]。因而,使催化中心富電子,如在Harbor-Bosch反應中加入電子促進劑如堿金屬、堿土金屬和稀土元素[24],或者在固氮酶中Fe蛋白向FeMo蛋白輔因子傳遞電子[8],則驅動活性中心向N2傳遞電子而促進分子氮活化。
半導體光催化中光生電子由催化劑價帶躍遷到導帶。以TiO2為例,光生電子由O的2p軌道躍遷到Ti的3d軌道中。以金屬原子d軌道吸附分子氮,則光催化劑在光生電子作用下還原N2而固氮。但是具有良好N2吸附能力的Ti,Fe元素,由于對應半導體(TiO2、Fe2O3)對N2的活化不足,它們的光催化固氮活性仍然十分有限。針對這一問題,作者團隊模擬固氮酶FeMo蛋白輔因子催化功能,提出以富電子氧空位為活性中心,促進N2在半導體光催化劑表面的吸附活化。氧空位是幾何結構的缺陷位(圖3a和3b),并包含局域電子的低價態金屬原子。以BiOBr為模型催化劑,通過密度泛函理論(density functional theory, DFT)計算發現BiOBr氧空位局域電子,幾何優化結果表明當分子氮靠近{001}面氧空位時,1個N原子占據氧空位以“end-on”形式吸附,通過化學鍵與周圍2個Bi原子配位而穩定存在。由于低價態Bi原子對N2反鍵軌道的電子注入,使分子氮中N—N鍵伸長為1.133 ?,鍵長介于N≡≡N雙鍵(1.201 ?)和三鍵(1.078 ?)之間,表明氧空位是良好的分子氮活化位點(圖3c和3d)[14]。由于氧空位能級處在導帶底下方,在光照下光生電子優先傳遞到氧空位,進而傳遞給氧空位所吸附的分子氮而還原N2。
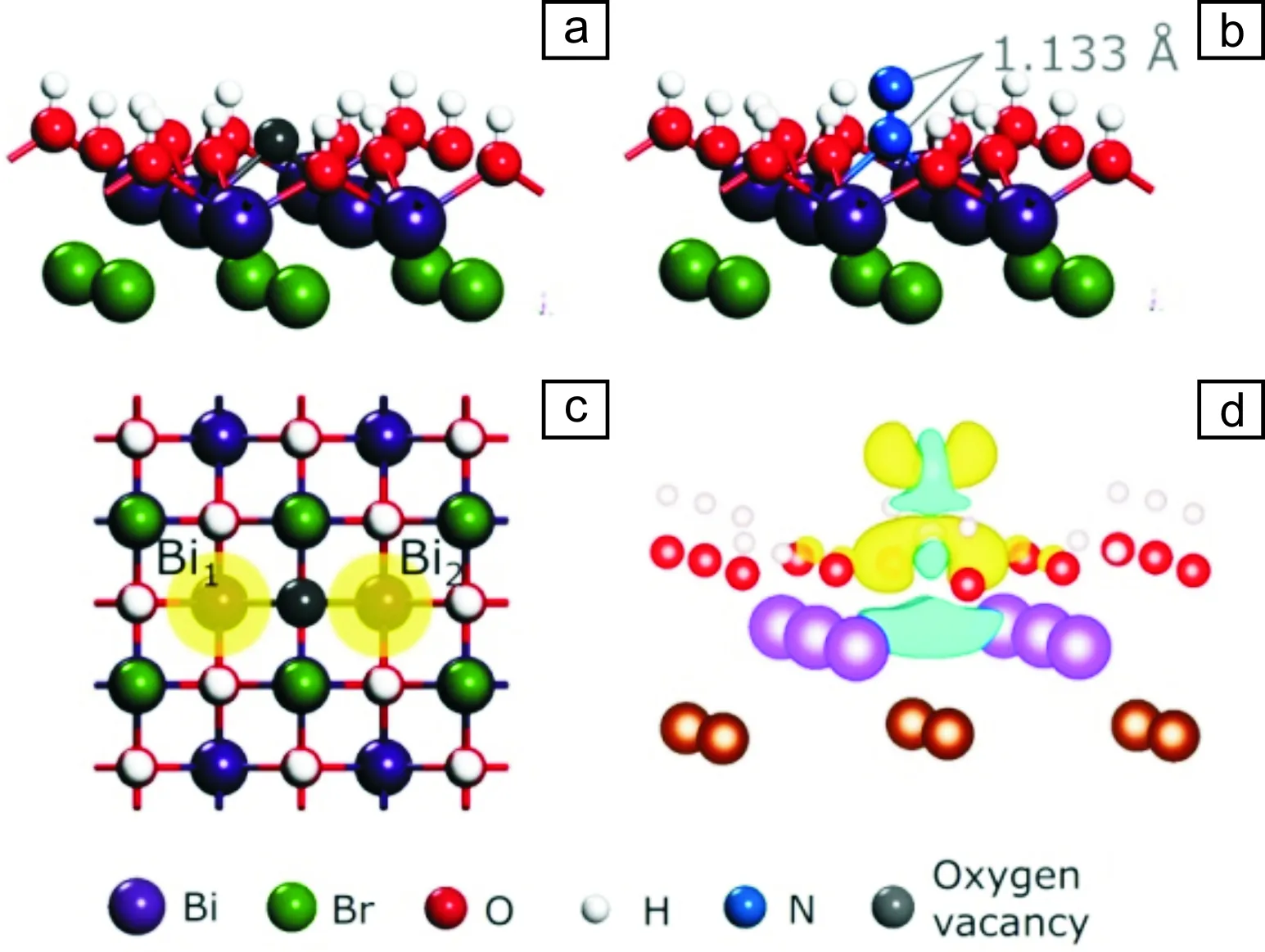
圖3 BiOBr{001}面氧空位結構:(a)側視圖, (b)俯視圖;N2在BiOBr{001}面氧空位的吸附構型(c)和對應差分電荷密度圖(d) [14]Fig.3 Side-view (a) and top-view (b) of OV structure on the {001} facet of BiOBr; N2 adsorption mode on the OV: (c) atomic structure and (d) corresponding charge difference[14]
氧空位因其周圍原子結構不同而具有不同的幾何結構和電子結構,進而影響其反應活性。作者團隊通過調控暴露晶面和氧空位濃度實現了BiOCl氧空位結構調控,并以含氧空位{001}和{010}晶面暴露BiOCl為模型催化劑,研究氧空位結構對分子氮活化的影響(圖4a)[25]。DFT理論計算結果表明,{001}晶面氧空位傾向于以“end-on”形式吸附N2,僅有1個N原子和2個Bi原子成鍵,而{010}晶面氧空位以“side-on”形式吸附N2,2個N原子分別與鄰近的3個Bi原子成鍵。由于2個N原子同時吸附活化,{010}晶面氧空位“side-on”N2吸附形式更大程度活化分子氮,將N—N鍵鍵長活化伸長到1.198 ?,而{001}晶面氧空位僅能活化分子氮到1.137 ?(圖4b和4c)。能量計算也表明{010}晶面氧空位更易吸附N2,其對N2吸附能比{001}晶面氧空位高出9.67 kcal/mol(圖4d)。這一結果與N2程序升溫脫附(N2-TPD)實驗結果吻合,BiOCl{010}晶面氧空位對N2吸附更強,因而其N2脫附溫度相對于{001}晶面氧空位高32 ℃(圖4e)。
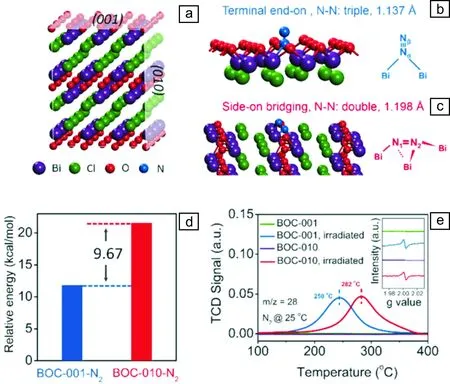
圖4 BiOCl原子結構模型(a), N2在BiOCl{001}晶面氧空位“end-on”吸附構型(b), N2在BiOCl{010}晶面氧空位“side-on”吸附構型(c), BiOCl{001}和{010}晶面氧空位對N2吸附能(d), {001}和{010}晶面暴露BiOCl樣品的N2程序升溫脫附(N2-TPD)結果(內置圖片為幾種樣品氧空位表征的電子順磁共振(EPR)譜)[25]Fig.4 Atomic structure of BiOCl (a), and N2 adsorption mode on the {001} OV (b) and {010} OV (c); Calculated energy difference of N2 adsorption (d); N2-TPD spectra of {001} and {010} exposed BiOCl (inset is the corresponding EPR spectra) [25]
大量光催化固氮工作表明,其他氧化物氧空位也能顯著提升催化活性。Dong等[26]報道Bi2MoO6氧空位周圍配位不飽和的Mo原子具有吸附活化N2能力,通過缺陷調控如還原暴露更多活性Mo原子可進一步增強Bi2MoO6固氮活性。Ye和Chen等[27]報道非化學計量比溴氧鉍Bi5O7Br超細納米管(直徑僅為5 nm)由于較寬的光譜吸收能力和大量氧空位暴露而成為優異的固氮材料。Zhang等[28]報道含氧空位的層狀雙氫氧化物MIIMIII-LDH (其中MII=Mg,Zn,Ni,Cu;MIII=Al,Cr),由于所制備材料厚度僅為幾個納米而暴露大量氧空位,多種不同金屬元素組成的LDH材料均可實現氧空位局域電子吸附活化N2,并且氧空位存在所誘導的晶格壓縮應力能進一步促進N2活化而有效固氮,其中CuCr-LDH活性最高。Shiraishi等[29]報道TiO2氧空位可活化分子氮,并且氧空位臨近Ti原子可提供兩個鈦羥基H供被活化的氮分子質子化而生成Ti-NH-NH-Ti,在光照下,水分子氧化而向Ti-NH-NH-Ti物種持續提供質子直至生成兩個Ti-NH3,從而實現N2到NH3的光化學轉換。
3.4 其他缺陷位介導的光催化固氮
近年來的研究結果表明,在非氧化物半導體如過渡金屬氮化物和硫化物中,催化劑固氮活性也表現出缺陷依賴特性。Wang和Dong等[30]報道g-C3N4由于空位缺失原子和待活化分子具有同種元素,因而N空位可選擇性吸附分子氮并傳遞空位剩余電子到分子氮,進而在光激發下活化和還原N2生成NH3。Wu等[31]制備的多孔海綿狀C3N4由于具備大比表面積、暴露充足N空位而具有較高光催化固氮活性。Liu等[32]也報道微波合成含N空位g-C3N4也具有光催化固氮效果。Hu等[33, 34]報道了三元金屬硫化物Zn0.1Sn0.1Cd0.8S和Mo0.1Ni0.1Cd0.8S中S空位是N2吸附活化中心,同時促進光生載流子分離,并且通過調控3種金屬配比而調控催化劑中S空位含量,發現催化劑固氮性能和S空位含量正相關。這些結果表明如S, N等缺陷同樣促進硫化物和氮化物材料光催化固氮性能。
雖然S, N, O缺陷缺失元素不同而導致缺陷化學差異,但三者同屬陰離子缺陷,在固氮機制中表現出相似性:S, N空位與O空位都具有剩余電子和原子級缺陷位,這樣一種富電子的缺陷結構為分子氮吸附活化提供位點;并且在能帶結構上,陰離子缺陷都在半導體導帶下方構建缺陷能級,當含這些陰離子缺陷的半導體受激發時,光生電子優先傳遞到缺陷能級而促進吸附態分子氮還原質子化。
4 增強氧空位光催化固氮效率策略
基于半導體缺陷位的光催化固氮活性研究成果如表1所示,氧空位作為一種新型光催化固氮活性位點顯著提高材料固氮活性,但一般材料氧空位濃度受限于其易猝滅和過高濃度氧空位引起晶體結構坍塌的特性,目前氧空位固氮材料活性仍然遠低于Harbor-Bosch法合成氨效率而不具有工業價值。其次由于光催化固氮材料對太陽能利用有限,光催化固氮全光譜理論量子效率(QE)或者光到氨(solar-to-ammonia, STA)的轉換效率仍然非常低,通常低于0.1%[18]。因而基于氧空位的光催化固氮效率仍有待提高。
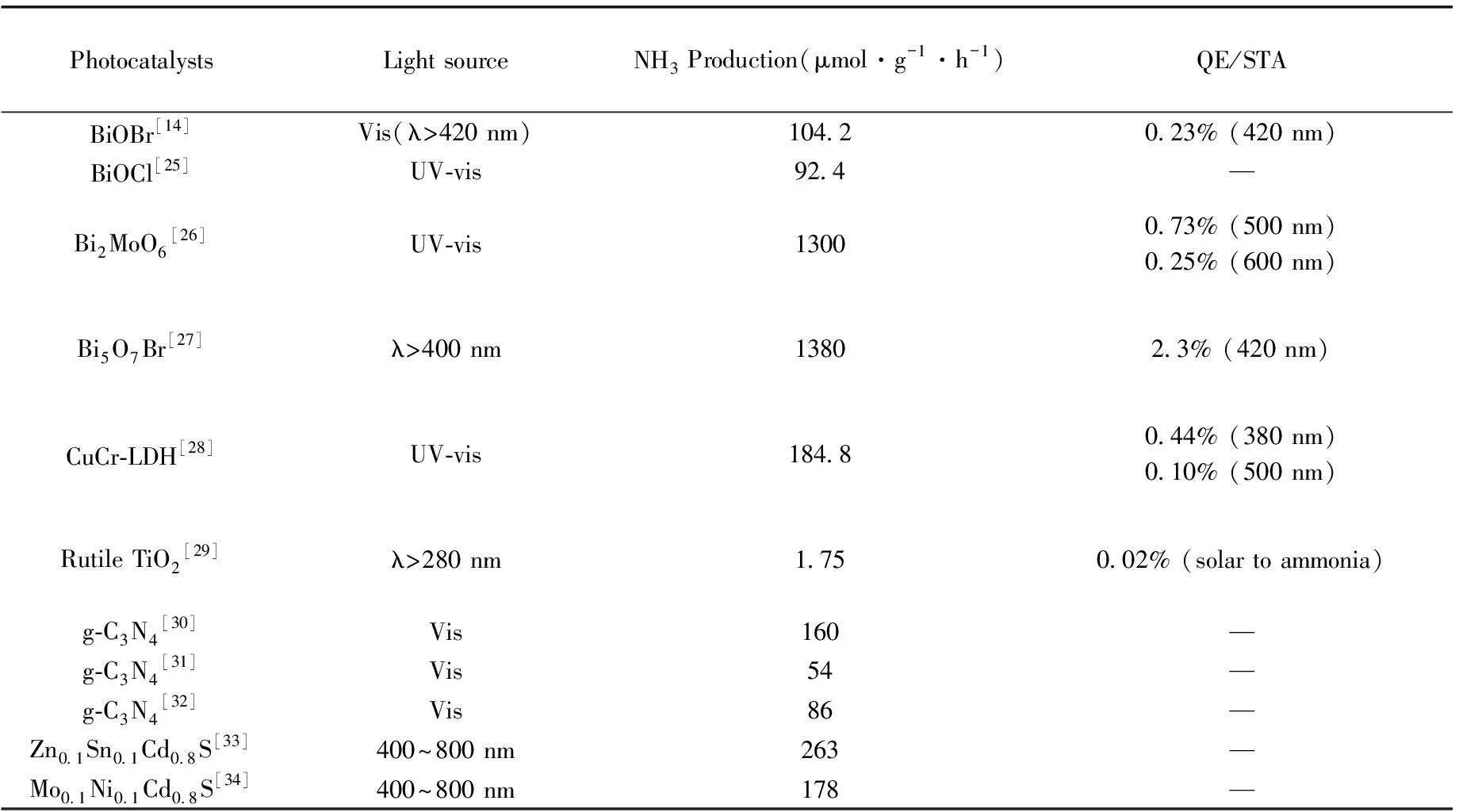
表1 基于半導體缺陷位的光催化固氮活性
4.1 異原子摻雜與半導體復合
Schrauzer等[9]報道Fe,Mo,Co,Ni摻雜TiO2促進其光催化固氮活性,但該文獻合成方法使這些金屬部分以金屬氧化物形式負載在TiO2上,形成兩種半導體催化劑的復合材料,表明異原子摻雜和多種光催化劑復合都可能促進光催化固氮活性,因而在氧空位基礎上引入異原子摻雜或半導體復合可促進氧空位基光催化劑活性。最近,Zhao等[35]的研究結果表明,過渡金屬元素如Fe摻雜在TiO2中引入氧空位,Fe活性中心和氧空位活性中心可能共同促進Fe摻雜TiO2光催化固氮活性。Hu和Li等[36, 37]報道g-C3N4/Zn0.12Mo0.12Cd0.9S1.14和g-C3N4/Zn0.11-Sn0.12Cd0.88S1.12復合材料中S空位為固氮活性中心,通過g-C3N4與三元金屬硫化物復合,促進光生載流子的分離而提高固氮活性。Xie和Xiao等[38]報道了在含缺陷二維C3N4表面引入Cu單原子,由于單原子Cu固定在C3N4的T缺陷位即C缺陷處,促進Cu單原子從C3N4共軛π電子云中提取電子進而吸附活化分子氮。
4.2 金屬納米晶
工業Harbor-Bosch法采用基于Fe, Ru催化劑合成氨,并發現其他過渡金屬如Os,Ni,Co等也具有催化合成氨活性,研究表明其活性中心大多為這些金屬的零價態納米晶,由于納米顆粒的小尺寸和特定晶面效應而暴露大量配位不飽和位點成為N2活化中心。此外,部分納米晶如Au, Ag, Cu的等離子共振效應引起了人們廣泛興趣,由于這些金屬在光照激發下產生熱力學非平衡熱電子隨光波做周期性振蕩運動,納米晶表面形成局域電磁場和熱場,這些光物理過程不僅增強材料光吸收、促進與之接觸的半導體光生載流子分離,并且熱電子可以越過高的化學反應勢壘而加速光催化反應效率。在光催化固氮反應中,Zeng等[39]報道了Os-Au催化劑在光照下可高效固氮,并證明Au的光致等離子效應可促進電子向Os傳遞進而加速Os固氮效率。Misawa等[40, 41]報道Au/SrTiO3/Ru和Au/Nb-SrTiO3/Zr/ZrOx具有光催化固氮
活性,Au納米晶由于等離子共振效應促進SrTiO3(Nb-SrTiO3)光生載流子分離、電子定向移動到固氮活性中心Ru和ZrOx,進而提升材料固氮性能。因而通過在含氧空位半導體表面負載納米晶顆粒成為一種可能的活性增強策略。
作者團隊以含氧空位無序氫化氧化鈦為載體負載Ru納米晶(平均尺寸2 nm),制備了高效合成氨催化劑(圖5a),發現無序氫化氧化鈦富氧空位具有遠超于晶態半導體的未配對電子數量,這些電子可通過Ru-TiO2-xHx界面Ru——Ti鍵傳遞給金屬Ru而使之富電子(圖5b),這一電子效應促使Ru通過π電子反饋作用更大程度活化分子氮。在光照下該催化劑通過廣譜光吸收特性和Ru-TiO2-xHx等離子雜化效應誘導的高效光熱轉換效率自升溫到360 ℃,以Harbor-Bosch熱催化原子態組裝機理高效合成氨(圖5c),并且不同于傳統熱催化,Ru等離子效應使Ru納米晶周圍產生局域電場和熱場(圖5d和5e),電場促進分子極化、熱場加速反應速率而降低光催化反應表觀活化能(圖5f),使K/Ru/TiO2-xHx光催化合成氨效率高于同溫度熱催化反應效率,并且高于Harbor-Bosch催化劑K/Ru/Al2O3和K/Ru/MgO的熱催化性能[15]。Gong等[42]報道以Au負載的含氧空位TiO2可實現有效光電固氮,通過原子層沉積技術制備富含表面氧空位的無定型TiO2作為N2吸附活化中心,體相無缺陷TiO2高效吸收利用紫外光,傳遞光生電子到表面富氧空位無定型TiO2固氮,Au等離子促進可見光的吸收利用且傳遞熱電子到表面無定型TiO2而提高氧空位固氮效率。
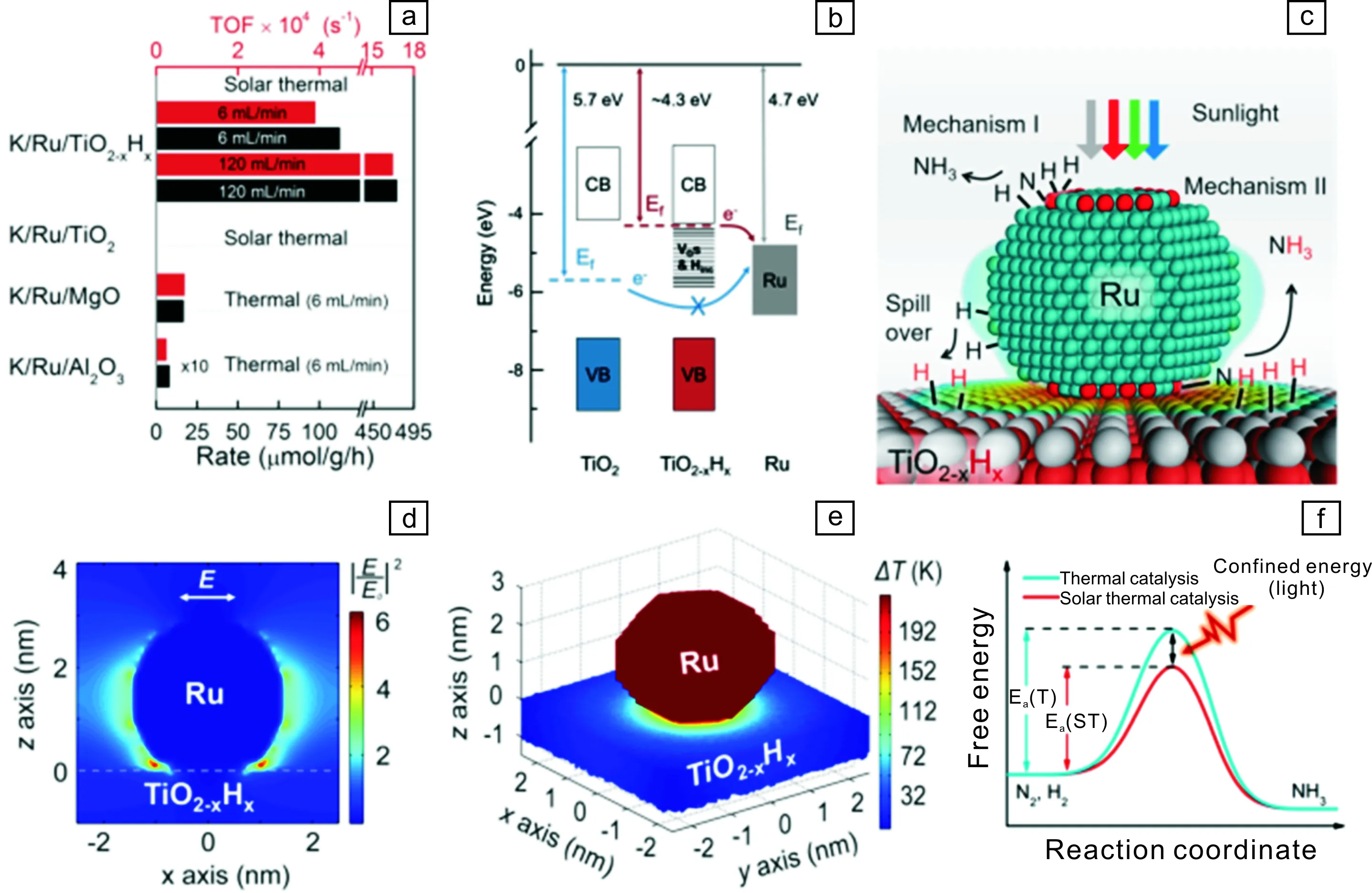
圖5 K/Ru/TiO2-xHx光催化合成氨效率 (a), Ru-TiO2-xHx電子傳遞示意圖 (b), K/Ru/TiO2-xHx光催化合成氨可能機理(c), 時域有限差分(finite-difference time-domain, FDTD)法模擬的Ru納米晶局域電場和熱場圖(d,e), K/Ru/TiO2-xHx光催化和熱催化合成氨表觀活化能對比(f) [15]Fig.5 The reactivity rate of ammonia synthesis of K/Ru/TiO2-xHx (a); Energy alignment of Ru, TiO2 and TiO2-xHx(b); Proposed mechanism of ammonia synthesis over TiO2-xHx supported Ru(c); Near field intensity(d) and heat (e)maps of TiO2-xHx supported Ru atomic, simulated using finite-difference time-domain (FDTD) method; The difference of Ea between thermal catalysis and solar thermal catalysis over K/Ru/TiO2-xHx[15]
5 結 語
本文通過歸納總結不同材料氧空位在固氮中的作用機制,表明氧空位配位不飽和金屬原子與局域未配對電子促進分子氮吸附活化是一種新型固氮活性位點。氧空位易制備、易調控,在不同氧化物固氮中有一定普適性,因而,基于氧空位的半導體光催化固氮活性中心設計具有應用潛力。
作為一種新型的活性中心,氧空位固氮方法的發展機遇與挑戰并存,主要包括:
(1)在水參與的光催化固氮反應中,氧空位傳導光生電子固氮為還原半反應,而對應空穴參與的半反應為極具挑戰性的水氧化反應(涉及多空穴傳遞而反應速度慢[43-45]),如何促進水氧化半反應可能是提高光催化固氮效率的潛在途徑。
(2)氧空位和固氮過渡金屬耦合是一種有潛力的固氮活性增強策略,氧空位的缺陷結構為過渡金屬均勻負載提供了錨定位點,并且其局域電子可能傳遞電子到過渡金屬d軌道而調控其固氮性能,尤其單原子催化[46, 47](極少量金屬單原子負載即可極大程度改變催化反應的活性和選擇性),為氧空位固氮活性增強帶來了機遇。
(3)光催化的低效率制約其發展。光催化反應如CO2還原、H2O分解、有機污染物降解和固氮的效率通常在μmol·g-1·h-1級別[48],而工業熱催化合成氨可達到mmol·g-1·h-1甚至更高,因而如何提高光催化固氮效率是亟待解決的關鍵問題。作者認為制備類似K/Ru/TiO2-xHx固氮作用機制的高效、廣譜光吸收催化劑,利用光熱效應實現高效光驅動合成氨是極具潛力的發展方向。
