銀杏葉提取物制劑中萜類內酯含量檢驗方法的建立
2019-04-01 11:45:52孔令偉時敬文胡金董官波
醫藥前沿 2019年4期
關鍵詞:方法
孔令偉 時敬文 胡金 董官波
(弘和制藥有限公司 吉林 梅河口 135000)
銀杏葉提取物制劑為靜脈注射劑,由銀杏葉提取物及部分輔料制成的滅菌水溶液,其主要成分為總黃酮、萜類內酯。其中總黃酮及萜類內酯為銀杏葉提取物的主要成分,因為中藥成分的復雜性,其質量標準均對主要成分的含量進行檢測,其中銀杏內酯具有抑制血小板聚集作用[1],對中風、老年癡呆癥等疾病具有明顯功效[2],因此其含量檢測方法成為內酯研究的熱點,本文通過使用高效液相色譜法檢驗銀杏內酯A、B、C、及白果內酯來指征銀杏總內酯的含量,并對該方法進行方法學驗證,結果表明該檢驗方法可用于產品的質量控制。
1.儀器和材料
Agilent 1260液相色譜儀,蒸發光散射檢測器(ELSD,Agilent公司),甲醇為色譜純(Fisher公司);梅特勒-托利多十萬分天平(型號MS105DU);其余試劑均為分析純。
銀杏葉提取物制劑為某制藥公司提供(批號:20170501,20170610);白果內酯(批號110865-201507)、銀杏內酯C(批號110864-201508)銀杏內酯A(批號110862-201612)、和銀杏內酯B(批號110863-201611)由中國食品藥品檢定研究院提供。
2.溶液制備
2.1 對照品溶液的制備:
取白果內酯、銀杏內酯C、銀杏內酯A、和銀杏內酯B對照品適量,用75%甲醇配成每1ml分別白果內酯、銀杏內酯C銀杏內酯A、和銀杏內酯B對照品0.2mg、0.3mg、0.3mg和0.1mg的混合對照品溶液。
2.2 供試品溶液的制備:
精密量取裝量項下供試品溶液30ml,置50ml具螺紋蓋離心管中,加入乙酸乙酯20ml,旋緊蓋子,混勻,打開,放氣,再旋緊蓋置渦旋振蕩器中振蕩1分鐘,離心(2000轉/分鐘)10分鐘,吸取乙酸乙酯置蒸發皿中,再加入乙酸乙酯5ml,重復操作5次,合并6次乙酸乙酯液,于80度水浴中蒸至近干,用4ml甲醇分次轉移至10ml離心管中,小心吹干甲醇,再精密加入75%甲醇0.5ml,渦旋使溶解,過0.22um的濾膜,取續濾液;或高速離心取上清液,即得。
3.色譜條件
Agilent ZOBAX SB-C18色譜柱(150mm×4.6mm,5μm)色譜柱,柱溫為35℃,以甲醇:0.1%甲酸(20∶80)為流動相進行洗脫。蒸發溫度為120℃,霧化溫度為90℃,載氣流速為1.1ml/min。進樣量:分別精密吸取對照品溶液3μl~10μl及供試品溶液5~10μl,注入液相色譜儀。
4.方法學考察
4.1 方法學摸索
4.1.1 由于每1ml制劑產品中僅含0.16~0.17mg銀杏葉提取物,推算其萜類內酯含量約在5~10μg/ml,含量較低,為達到檢測靈敏度,需要從檢測和供試品前處理兩方面均考慮,盡量提高檢測靈敏度,滿足含量測定的定量要求。
4.1.2 色譜條件的確定
(1)蒸發光散射檢測參數
研究發現,蒸發光檢測器的蒸發溫度和霧化溫度越高,4種待測成分的響應就越高;待測成分的響應隨著氣體流速的降低而升高,至1.1ml/min時,響應最高[3]。
最終蒸發光散射檢測器確定參數為:霧化溫度90℃,蒸發溫度為120℃,氣體流速為1.1ml/min。由于注射液中萜類內酯含量低,如檢測器靈敏度不夠可能導致無法檢出,因此在標準中細化蒸發光檢測器參數作為參考。
(2)流動相酸度
研究發現,流動相具有一定酸度會增強萜類內酯信號,考慮到蒸發光散射器特性,選擇揮發性酸甲酸進行考察,比較水、0.01%甲酸、0.1%甲酸和0.5%甲酸作為水相與甲醇洗脫時,發現0.1%甲酸為流動相水相時響應最強,因此選擇流動相為0.1%甲酸-甲醇(80∶20)。
確定色譜條件下,4種萜類內酯分離較好,供試品中無雜峰干擾4種待測成分(見圖1)。
4.1.3 供試品前處理方法摸索 分別摸索了取大體積注射液,進行濃縮后再液液萃取的方法(方法1和方法2,同時比較不同溶劑提取效率)、直接取少量注射液進行渦旋萃取(方法3)、固相萃取方法(方法4)和直接液液萃取方法(方法5),具體方法如下。
(1)方法1:精密吸取裝量項下供試品溶液300ml,至500ml圓底燒瓶中減壓濃縮至近干,加無水乙醇100ml溶解殘渣,過濾,取濾液至蒸發皿中,于水浴蒸至約2ml 轉移至50ml具螺紋蓋的離心管中,用無水乙醇2ml洗滌蒸發皿轉移至同一離心管中,再量取水10ml清洗圓底燒瓶同樣轉移至同一離心管中,用乙醚約20ml清洗上述蒸發皿,轉移至同一離心管中,旋緊蓋子后混勻,打開蓋放氣,再旋緊蓋置至渦旋振蕩器中振蕩1分鐘,離心(3000轉/分鐘)5分鐘,分取上層乙醚液體至蒸發皿中,再加入乙醚20ml,按照上述提取步驟提取,分離乙醚,重復操作4次,合并5次提取的乙醚液,室溫揮至近干,加甲醇定容至5ml。

圖1 注射液中萜類內酯含量測定色譜圖
(2)方法2:精密吸取裝量項下供試品溶液300ml,至500ml圓底燒瓶中減壓濃縮至近干,加無水乙醇100ml溶解殘渣,過濾,取濾液至蒸發皿中,于水浴蒸至約2ml轉移至50ml具螺紋蓋的離心管中,用無水乙醇2ml洗滌蒸發皿轉移至同一離心管中,再用水10ml清洗圓底燒瓶同樣轉移至同一離心管中,用乙酸乙酯約20ml清洗上述蒸發皿,轉移至同一離心管中,旋緊蓋子后混勻,打開蓋放氣,再旋緊蓋置至渦旋振蕩器中振蕩1分鐘,離心(3000轉/分鐘)5分鐘,分取上層乙酸乙酯液體至蒸發皿中,再加入乙酸乙酯20ml,按照上述提取步驟提取,分離乙酸乙酯,重復操作4次,合并5次提取的乙酸乙酯液,室溫揮至近干,加甲醇定容至5ml。
(3)方法3:精密吸取裝量項下供試品溶液30ml,置50ml具螺紋蓋的離心管中,加25%鹽酸2滴,加入乙酸乙酯10ml,旋緊蓋子后混勻,打開蓋放氣,再旋緊蓋置渦旋振蕩器中振蕩1min,離心(3000轉/min)5分鐘,取上層乙酸乙酯置蒸發皿中,再加入乙酸乙酯5ml,按上述提取步驟提取,分離乙酸乙酯,重復操作4次,合并5次提取的乙酸乙酯液,置80度水浴中蒸至近干,用3ml甲醇分次洗脫至10ml離心管中,吹干甲醇后,再精密加入50%甲醇0.5ml,渦旋使溶解,離心,取上清液,即得。
(4)方法4∶精密吸取裝量項下供試品溶液50ml,過經活化后的固相萃取柱(Waters HLB 6CC,使用前加6ml甲醇過柱,棄去甲醇,再加6ml水過柱,棄去水進行活化;流速:1滴/秒;),棄去已過柱的供試品,量取乙酸乙酯6ml洗脫固相萃取柱,收集乙酸乙酯洗脫液,至10ml具螺紋蓋的離心管中,吹干,精密加入甲醇1ml,渦旋振蕩溶解,離心,取上清液,即得。
(5)方法5:精密吸取裝量項下供試品溶液100ml,量取乙酸乙酯40ml萃取,分取上層乙酸乙酯液體至蒸發皿中,再加入乙酸乙酯30ml按照上述提取步驟提取,分離乙酸乙酯,重復操作3次,合并4次提取的乙酸乙酯液,(每次萃取時將乳化的上層轉移至50ml具螺紋蓋的離心管中,離心,再吸取乙酸乙酯至蒸發皿中),置水浴上揮至近干,加3ml甲醇分次清洗蒸發皿,轉移至離心管內,吹干,殘渣加甲醇定容至1ml。
取上述不同前處理方法制備的供試品溶液,按照既定的色譜條件進樣分析,結果見表。
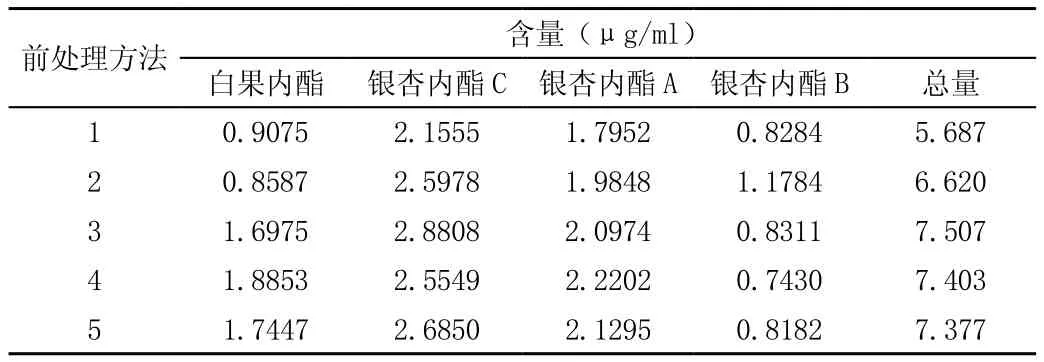
表 不同前處理方法萜類內酯結果
結果表明:(1)大體積供試品進行蒸干后提取,供試品中白果內酯含量大大降低,可能在蒸干過程中損失;(2)方法1和和方法2分別采用乙醚和乙酸乙酯,結果表明乙酸乙酯提取效率高;(3)采用固相萃取與渦旋提取兩種方法,提取效率最高;(4)研究中按照方法4固相萃取法采用另一家品牌的固相萃取柱(愛捷爾)進行前處理,結果均未檢出4種萜類內酯,方法耐用性受色譜柱品牌所限。
綜上,渦旋提取法提取效率高,且操作簡便,快速,故選用該法作為前處理方法,銀杏葉提取物制劑中萜類內酯含量測定檢驗方法可用于銀杏葉提取物制劑萜類內酯含量的質量控制,該方法線性在0.9994~0.9999范圍內,準確性、靈敏度均較高。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
