葛根素與水二聚體相互作用的密度泛函理論研究
2019-04-11 06:45:30
中國民族民間醫藥 2019年4期
廣東藥科大學中藥學院,廣東 廣州 510006
葛根素(Puerarin)來源于豆科植物野葛(PuerarinLobata(Willd.)Ohwi)或甘葛藤(PuerarinthomsoniiBenth.)的干燥根中提取出的一種異黃酮苷[1],分子式為C21H20O9,相對分子質量為416.38,呈白色針狀結晶。其毒性小、安全范圍廣、療效好,臨床上主要用于心腦血管疾病的治療[2]。但其水溶性較差限制了葛根素在臨床上的廣泛使用[3]。葛根素具有異黃酮類物質化學結構的特點,β環受吡喃環羰基的立體阻礙影響,在空間上只能形成大的共扼體系而成為非平面結構,且晶格排列緊密,剛性較強[4]。葛根素在8位上連一吡喃葡萄糖,在7,4位上各有一羥基,為葛根素的活性基團。其苷元水溶性很差,因此認為葡萄糖基是葛根素有一定水溶性的重要因素。而結構中的羥基(-OH)均有可能和水分子形成分子間氫鍵,所以本研究設計了6種C21H20O9-H2O超分子體系的結構,通過比較分子之間的相互作用強弱,來揭示相互作用的本質,對了解葛根素與水的作用方式有重要意義[5]。
1 計算方法
分子模擬的主要方法有經典力學模擬和量子力學模擬,而Gaussian一般適用于量子力學計算。其中量子力學模擬主要依據的方法有從頭計算法、半經驗分子軌道理論、密度泛函理論(DFT)方法等。而密度泛函方法是一個較好的理論計算和結構預測方法,對于確定復雜結構具有重要的參考意義[6]。本文以Gview 3.7軟件組建葛根素單體C21H20O9和H2O的二聚體C21H20O9-H2O,計算方法均使用DET-B3LYP/6-31+G方法。其中,B3LYP是密度泛函理論中常用的方法之一,6-31G是描述原子的基組,“6”是指對內層軌道的描述;“31”是指每個價層電子會被劈裂成2個基函數,分別由3個和1個高斯型函數線性組合而成;“G”是高斯基組。以本征值跟蹤算法(EF)和內坐標的Berny算法等[7]對所選的結構進行全優化,求得能量極小點(穩定構型),經振動分析得優化結構無虛頻。采用Boys和Bernardi提出的均衡校正法(CP)[8]進行基組疊加誤差(BSSE)校正與零點能(ZPE)校正。通過自然軌道分析[9],揭示單體轉變為二聚體后的電荷轉移狀況,探索C21H20O9和H2O形成二聚體時的相互作用的本質。通過統計熱力學方法求得熱力學函數[10],揭示單體轉變為二聚體后的能量變化,探索C21H20O9在溶解過程的變化規律及反應機制。全部計算均采用Guassian 09W程序[11]完成,收斂精度取程序內定值。
2 結果與討論
2.1 幾何構型 圖1和圖2列出了優化后的葛根素和水分子的及原子編號,二聚體(Ⅰ~Ⅵ)的DFT-B3LYP/6-31+G全優化幾何構型部分幾何參數列于表1。其中酚羥基O25-H26(即葛根素異黃酮母核B環上的酚羥基)與水形成的二聚體稱為二聚體Ⅰ;酚羥基O27-H28(即葛根素異黃酮母核A環羥基)與水形成的二聚體稱為二聚體Ⅱ;羥基O44-H45與水形成的二聚體稱為二聚體Ⅲ;羥基O34-H35與水形成的二聚體稱為二聚體Ⅳ;羥基O36-H37與水形成的二聚體Ⅴ;羥基O41-H42(即葡萄糖五元環結構所連亞甲基上的羥基)于水形成的二聚體稱為二聚體Ⅵ。
由表1可見,二聚體(Ⅰ~Ⅵ)中的接觸點(O-H…O)距離分別為0.169 2、0.258 9、0.158 0、0.160 6、0.171 5和0.309 4 nm,長短次序為:Ⅲ<Ⅳ<Ⅰ<Ⅴ<Ⅱ<Ⅵ,接觸點距離最短屬于二聚體Ⅲ。二聚體(Ⅰ~Ⅵ)均近似地屬于C1點群,與單體相比,二聚體(Ⅰ~Ⅵ)接觸點為O-H…O的O-H鍵長變化分別為0.001 8、0.001 2、0.002 7、0.002 6、0.001 7和0.000 9 nm,二聚體Ⅲ的O-H鍵長變化最大。與單體相比,二聚體鍵角變化均小于0.8°,二面角變化均小于0.2°。由此可見,聚合和分子間相互作用未造成各單體較大的扭曲,對內旋轉影響也比較小。根據二聚體的接觸點種類和距離可以初步推測出結果:二聚體Ⅲ的穩定性最優。


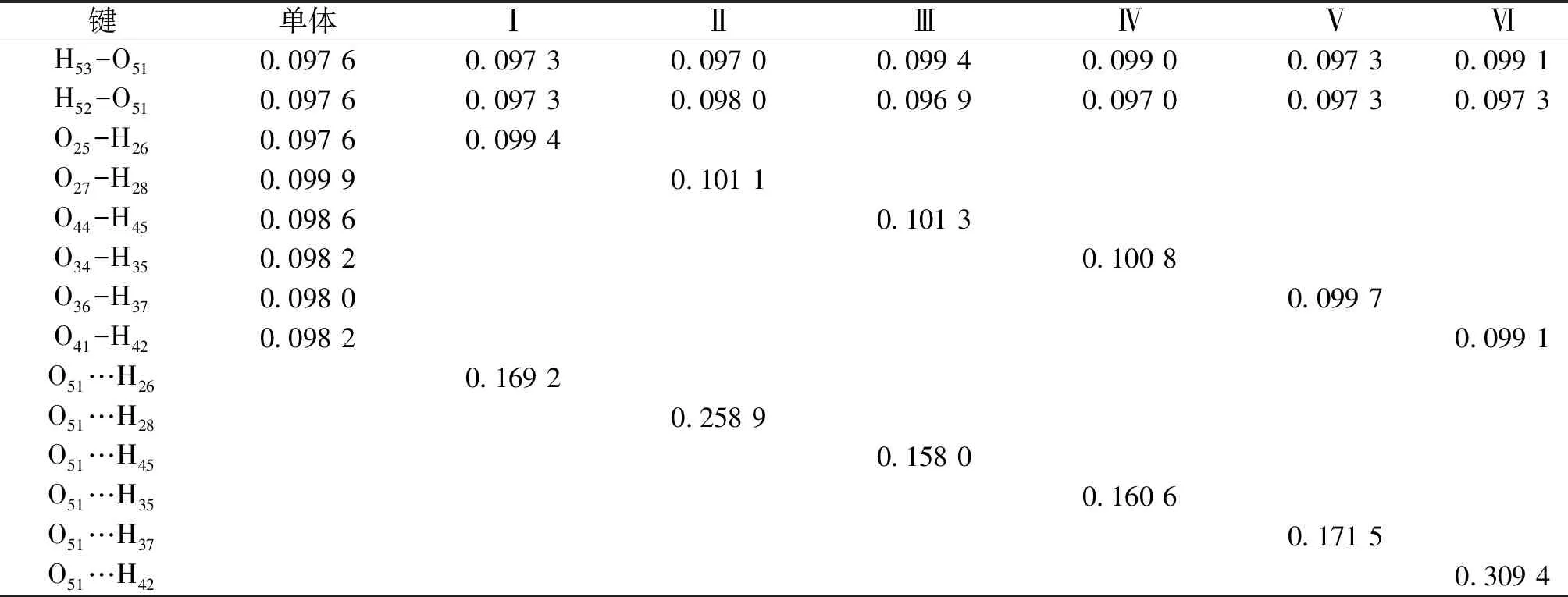
表1 葛根素,水和二聚體(Ⅰ~Ⅵ)的部分優化鍵長 (nm)
2.2 電荷分布及轉移 由表2可見,與單體相比,在接觸點上的氧原子,氫原子及附近的氧原子變化明顯。如二聚體Ⅲ中的O44為-0.817 e減少了0.027 e,H45為0.540 e增加了0.011 e。結合后H2O上的氧原子和氫原子的電荷也有明顯變化。子體系間電荷轉移比較多。在二聚體(Ⅰ~Ⅵ)中兩子體系間電荷傳遞主要通過接觸點O-H…O傳遞電荷分別為0.051、0.028、0.029、0.028、0.052和0.022e。

表2 葛根素,水和二聚體(Ⅰ~Ⅵ)的部分優化自然原子電荷 (e)
2.3 結合能 由表3可知,在DFT-B3LYP/6-31+G水平下求得的葛根素和水形成二聚體時的分子間相互作用能,有總能量(E)和經基組疊加誤差(BSSE)和ZPE校正后的結合能(ΔEc)兩部分。二聚體(Ⅰ~Ⅵ)結合能分別大小順序為:Ⅲ﹥Ⅳ﹥Ⅵ﹥Ⅴ﹥Ⅰ﹥Ⅱ。經BSSE和ZPE校正后的葛根素與水二聚體的最大結合能為-79.82 kJ·mol-1,屬于構型Ⅲ,而二聚體Ⅱ的結合能較低,這是由于所連接的酚羥基受葡萄糖糖基的位阻影響,活性較弱,這與前面的穩定化能及根據鍵長和接觸點距離來推測二聚體穩定性基本相一致。
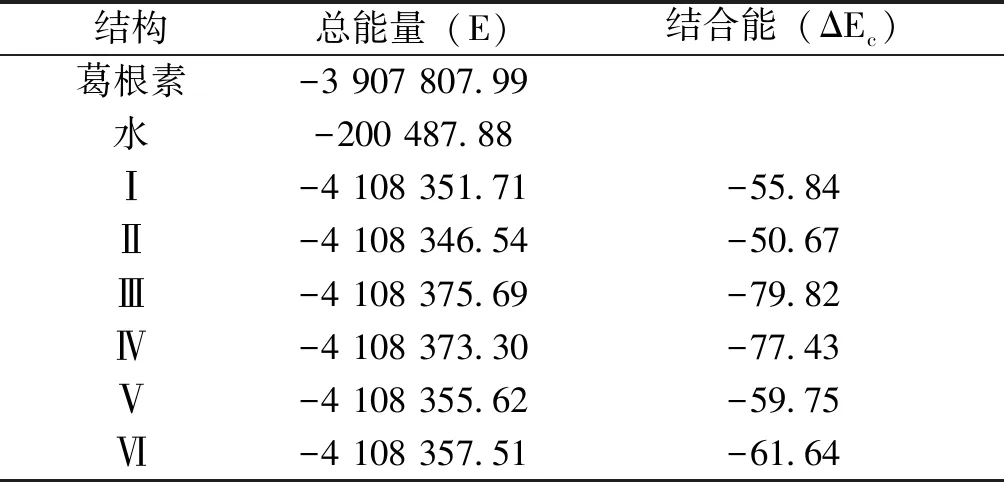
表3 葛根素,水和二聚體(Ⅰ~Ⅵ)總能量及結合能 (kJ·mol-1)
2.4 熱力學性質 基于統計熱力學的方法,在振動分析基礎上(頻率校正因子為0.96[10]),計算在298.15 K下由單體生成二聚體(Ⅰ~Ⅵ)的熱力學函數的變化值,見表4。由單體生成二聚體時,體系熵值減小,分子間相互作用使聚合過程焓值變大,是吸熱過程,形成二聚體(Ⅰ~Ⅵ)焓變值Ⅲ﹥Ⅳ﹥Ⅴ﹥Ⅵ﹥Ⅱ﹥Ⅰ,表明二聚體Ⅲ,Ⅳ,Ⅴ,Ⅵ,Ⅱ,Ⅰ分子間相互作用依次減弱。分子間相互作用最強的二聚體為Ⅲ,這與前面推論一致。

表4 298.15 K下由單體形成二聚體的熱力學性質
注:△ST=(ST)dimer-2(ST)monomer,△HT=(HT+E(HF)+ZPE)dimer-∑(HT+E(HF)+ZPE)monomer
3 結論
由葛根素結構分析,黃酮母核上的有兩個酚羥基,由于酚羥基與苯環已形成了共軛體系,很難像醇羥基一樣發生反應,故酚羥基上與水分子發生氫鍵作用形成二聚體的穩定性可能較差。因而連接在葡萄糖苷上的4個羥基與水相互作用形成的二聚體穩定性可能較優。
在DFT-B3LYP/6-31+G水平下,求得的葛根素與水二聚體的優化結構(Ⅰ~Ⅵ)。而且經振動分析得優化結構無虛頻,具有穩定的結構。與單體相比,二聚體(Ⅰ~Ⅵ)接觸點為O-H…O的O-H鍵長均變大,接觸點距離最小的屬于構型Ⅲ。而且各接觸點上的氧原子,氫原子及附近的氧原子電荷變化明顯。對葛根素與水形成二聚體(Ⅰ~Ⅵ)的自然鍵軌道,相互作用能和熱力學性質上來分析,最大的結合能和焓變值分別為79.82 kJ·mol-1和-20.92 kJ·mol-1,均屬于構型Ⅲ。因此,二聚體Ⅲ為最穩定結構。
