慢性發病的皮下脂膜炎樣T細胞淋巴瘤一例
2019-05-05 10:51:52徐麗萍宋欣偉關天容
浙江中西醫結合雜志 2019年4期
吳 廳 徐麗萍 宋欣偉關天容
1 病歷簡介
患者,男,47歲,因四肢軀干多發腫塊1年余,下肢紅斑水泡4天于2018年2月19日入住浙江省中醫院風濕免疫科。患者2016年6月開始無明顯誘因出現左肘部腫塊,質硬,無壓痛,逐漸增大至5cm×6cm左右,當地醫院行腫塊組織活檢,提示橫紋肌軟組織內肉芽腫性炎伴散在灶性壞死,并伴大量慢性炎細胞浸潤,考慮感染性病變,特殊染色未發現細菌或微生物。2017年3月左肘腫塊進一步增大,并出現四肢、軀干多發腫塊,當地醫院查左肘增強MRI提示感染性病變可能,韌帶樣纖維瘤待排。并赴上海某醫院再行腫塊組織活檢,提示橫紋肌組織內大量炎癥細胞浸潤,芽腫性炎伴大片壞死,炎癥細胞見大量嗜酸細胞和漿細胞,另見血管炎,考慮免疫性疾病。2017年8月四肢軀干腫塊逐漸增多增大,多地就診診斷不明。4天前因全身多發紅斑伴散在水泡就診我院。入院后體格檢查:體溫38.2℃,心率113次/分,血壓 142/89mmHg(1mmHg=0.13328kPa),神清,精神尚可,淺表淋巴結未及明顯腫大,雙肘伸側、左腕屈側、背部、腹部、雙大腿內側多發腫塊,邊界尚清、質硬、無壓痛,局部皮溫、皮色正常。全身多發紅斑伴散在水泡,紅斑界限清楚,壓之褪色,水泡約1cm×1cm大小,部分破潰結痂。實驗室檢查:超敏C反應蛋白146.91mg/L。紅細胞沉降率12mm/H。血常規:白細胞計數3.2×109/L,單核細胞百分數10.6%,血紅蛋白132g/L,血小板計數224×109/L。生化類:堿性磷酸酶(ALP)85U/L,乳酸脫氫酶250U/L。EB病毒抗體IgM、IgG:陽性。EB 病毒 DNA:4.26×105/拷貝數。左肘增強MRI:左上臂團塊狀異常信號伴前、后方部分肌群及肱骨遠端、尺骨鷹嘴受累(見插頁圖1-2)。腹部皮膚及腫塊組織高通量病原體基因測序:EB病毒檢測人類皰疹病毒4型:2473。腿部皮膚及腫塊組織病理(見插頁圖3-4):真皮及皮下脂肪組織內見異型淋巴細胞浸潤伴大片壞死退變,免疫組化:異型淋巴細胞CD3(+)、CD4(-)、CD8(+)、CD7(-)、CD2(+)、CD5(少數+)、CD56(+)、CD30(+)、TIA-1(+)、GB(+),組織細胞 CD68/kp1(+)、Ki-67(約 70%+)。結合形態、免疫表型和分子生物學檢測結果,符合外周T和NK細胞腫瘤,較傾向NK/T性淋巴瘤。TCR基因重排:TCRG:A(+)B(-),TCRB:B(+)C(-),TCRD:A(-)。診斷:皮下脂膜炎樣T細胞淋巴瘤。于3月27日行PGemox方案(澤菲1.6g d1+奈達鉑 160mg d1+DXM10mg d1-4+培門冬37500U d5)化療,期間出現房顫、IV度骨髓抑制伴腹瀉、發熱、凝血障礙、十二指腸穿孔,經對癥處理后緩解,但患者及家屬放棄治療,于4月18日自動出院。
2 討論
皮下脂膜炎樣T細胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma,SPTCL),是一種罕見的以累及皮下脂肪組織為主的NK/T細胞淋巴瘤[1]。其中α/βT細胞淋巴瘤患者多表達CD8,病變多局限于皮下組織,無表皮或真皮的累及,其病程相對較長,很少伴有結節外病變或噬血細胞綜合征(hemophagocytic syndrome,HPS)。而 γ/δT 細胞淋巴瘤患者多表達 CD56,而 CD4(-),CD8(-),病變除了累及皮下組織外,還可出現表皮或真皮的侵犯,常伴有HPS,病程短、進展快,預后較差[2]。但本例患者病理組織免疫組化 CD4(-)、CD8(+)、CD56(+),病變累及全皮膚層及肌肉組織,病程長達1年余,進展緩慢,期間無發熱、乏力、消瘦等全身癥狀,看似惰性淋巴瘤表現,但后期進展快,化療效果不佳,不良反應多,預后差,臨床診治對此不能掉以輕心。SPTCL好發于中青年,無顯著性別差異,其發病原因不明,目前普遍認為與EB病毒感染有一定相關性,正如本例患者血清抗體、DNA及組織高通量病原體基因測序均提示EB病毒感染依據。
SPTCL的診斷有賴于病理檢查,其組織病理學特征類似于脂膜炎,主要有皮下脂肪組織內大小不一的非典型性淋巴細胞浸潤,并圍繞單個脂肪細胞呈花環樣排列,常見核分裂、脂肪組織壞死。組織細胞吞噬現象及凋亡小體,并可伴有血管浸潤和肉芽腫樣改變,免疫組化主要表達T淋巴細胞相關抗原,如CD45、CD45RO、CD3,部分患者CD8陽性,少數CD4陽性,也可表達CD2、CD43、CD56,也有少量腫瘤細胞表達CD5、CD7、CD30[3]。SPTCL 早期多表現為反復發作的無痛性皮膚結節,缺乏特異性,易被誤診。臨床上應注意與以下疾病相鑒別:良性脂膜炎:SPTCL早期皮膚改變可能和良性脂膜炎無異,SPTCL在組織病理學上也有脂膜炎和豆袋細胞,但瘤細胞有異形性、表達T細胞表型和細胞毒顆粒相關蛋白,與結節性脂膜炎不同;皮膚感染性疾病:也可出現皮膚結節,紅腫熱痛、潰瘍、分泌物增多,但本病對抗感染效果顯著;結節性紅斑:以皮膚血管炎和脂膜炎為病理基礎,組織學改變主要在皮下組織,真皮中只表現為血管周圍中等數量的慢性炎癥細胞浸潤,一般不會破潰,預后良好。本例患者既往體健,且發病1年內行兩次病理活檢均提示炎性改變,這可能是被誤診為感染的主要原因,入住我院后因皮膚破潰伴發熱,病理活檢有類似血管炎改變而按照免疫性疾病診治,雖皮膚腫塊、破潰癥狀有所好轉,但病情并未得到完全改善,聯系本院皮膚科和血液科多次會診建議取不同部位組織多次活檢最終依據免疫組化結果診斷為此病。這可能說明病理組化檢測到陽性結果需疾病進展至一定程度,北京醫科大學聯合醫院風濕病科曾報道8例SPTCL平均每例病理檢查2.75次才檢測到確診依據為病變部位下脂肪中有密集的淋巴細胞浸潤伴有明顯的異型細胞和CD3陽性[4],所以當證據不足時密切隨訪是明確診斷的重要方式。SPTCL發病率低,臨床罕見,目前仍無統一標準的治療方案,一般多采用CHOP(環磷酰胺+阿霉素+長春新堿+強的松)方案,但療效欠理想,本例采用P-Gemox方案,患者腫塊、紅斑較前有所消退,但期間出現多種不良反應及并發癥,目前已自動出院,電話隨訪所知患者目前狀況不佳。此例已是我科室收治的以疑似風濕病癥狀為首發表現,最終確診為淋巴瘤的第6例患者,其中SPTCL占到3例,分別表現為口腔潰瘍、發熱、皮膚腫塊,因其預后差,治療方案欠佳,故臨床上仍需積累更多經驗認識并治療該病,尤其是首診于風濕免疫科,擬診為皮膚腫塊、血管炎、白塞病、發熱原因不明的患者,應給予高度重視,多次病理活檢檢查雖然不易被患者及家屬接受,但病情需要時仍是無可替代的手段,同時,二代基因測序手段有助于疾病診斷,值得開展。總之,盡可能的及早診斷,及早治療,有助于提高生存率。

圖1 左肘后軟組織腫塊T1W稍高信號
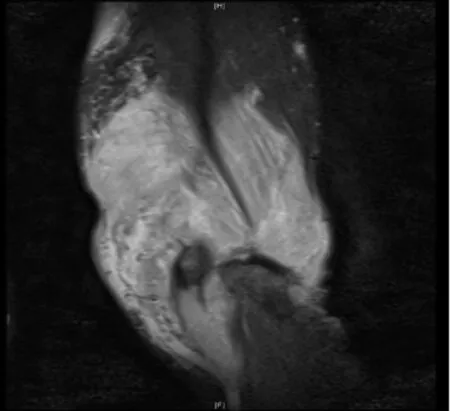
圖2 左肘后軟組織腫塊T2W高信號
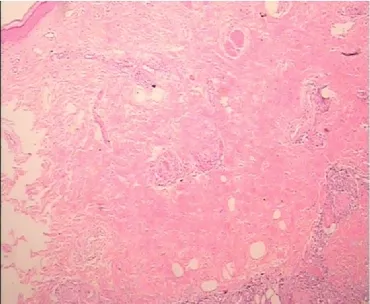
圖3 真皮及皮下脂肪組織內見異型淋巴細胞浸潤伴大片壞死退變(HE×100)
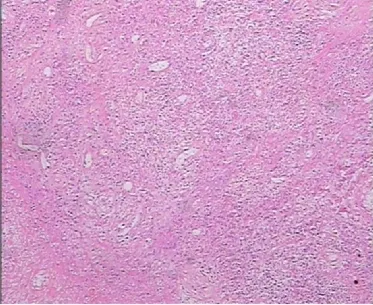
圖4 異型淋巴細胞浸潤(HE×400)
