含4 種食源性病毒檢測靶標多聯(lián)裝甲RNA的制備、純化與定值
2019-05-21 11:59:50李風鈴逄鳳嬌江艷華王聯(lián)珠翟毓秀
食品科學 2019年8期
關(guān)鍵詞:檢測
姚 琳,張 奇,李風鈴,張 媛,逄鳳嬌,江艷華,*,王聯(lián)珠,翟毓秀
(1.中國水產(chǎn)科學研究院黃海水產(chǎn)研究所,農(nóng)業(yè)部水產(chǎn)品質(zhì)量安全檢測與評價重點實驗室,山東 青島 266071;2.獐子島集團股份有限公司,遼寧 大連 116001)
諾如病毒(norovirus,NoV)、甲肝病毒(hepatitis A virus,HAV)、輪狀病毒(rotavirus,RV)、星狀病毒(astrovirus,AstV)等食源性病毒引發(fā)的公共衛(wèi)生與食品安全問題已成為影響全球人類健康的主要問題[1-2]。2006—2007年,日本、英國爆發(fā)NoV疫情,造成共約500萬 人感染;1988年我國上海爆發(fā)HAV疫情,造成約30萬 人感染;RV是嬰幼兒腹瀉的主要病原,世界范圍內(nèi)每年估計約有5%的兒童死于RV感染[3];目前AstV在一些國家的調(diào)查中發(fā)現(xiàn)已相當普遍,陽性檢出率在2%~8%之間[4-5]。食源性病毒可通過受污染的水、貝類、果蔬等引起疾病的爆發(fā)和流行[6-7]。實時熒光定量逆轉(zhuǎn)錄-聚合酶鏈式反應(reverse transcription polymerase chain reaction,RT-PCR)是檢測食品中食源性病毒的最主要方法。由于食品基質(zhì)復雜、干擾因素多且病毒含量較低,通常需要在檢測過程中添加陽性質(zhì)控樣品,以確保檢測結(jié)果的準確性和可靠性。對于上述RNA病毒,近年來有研究者采用體外轉(zhuǎn)錄的裸露RNA片段作為實時熒光定量RT-PCR質(zhì)控樣品[8-10],但存在不能監(jiān)控病毒粒子裂解、RNA提取等關(guān)鍵環(huán)節(jié)且易降解等缺點。
針對上述問題,Dubois等[11]建立了裝甲RNA技術(shù),即通過分子生物學手段將目標RNA片段包裹在MS2噬菌體病毒樣顆粒(virus like particles,VLPs)內(nèi)部,使之能抵抗環(huán)境和樣本中核酸酶的降解,同時其整體結(jié)構(gòu)又很好地模擬了病毒衣殼蛋白-核酸天然存在的特征,具有穩(wěn)定、無生物安全隱患、可有效評價病毒裂解與提取效率等檢測全過程的優(yōu)點,該技術(shù)已成功應用于腸道病毒[12]、人體免疫缺陷病毒[13]等病毒的陽性質(zhì)控樣品的制備。然而,目前鮮見可同時適于多種病毒檢測陽性質(zhì)控樣品的研究報道。
本研究在前期構(gòu)建基于Qβ噬菌體的裝甲RNA制備平臺的基礎(chǔ)上[14],針對食源性病毒的主要檢測標準,研究并純化了同時包含NoV、HAV、RV、AstV 4 種食源性病毒檢測靶標的多聯(lián)裝甲RNA,并對其進行初步定值,為研發(fā)用于食源性病毒核酸檢測的多聯(lián)陽性質(zhì)控樣品提供新思路。
1 材料與方法
1.1 材料與試劑
大腸桿菌BL21(DE3)和Top10感受態(tài)細胞 天根生化科技(北京)有限公司;pET-28a(+)載體為本實驗室保存;pGEM-T-Easy載體、T7 RiboMAXTMExpress Large Scale RNA Production System 美國Promega公司;同源重組克隆試劑盒ClonExpress?II One Step Cloning Kit 南京諾唯贊生物科技有限公司;限制性內(nèi)切酶、Premix Ex TaqTM、One Step PrimeScriptTMRT-PCR Kit 寶生物工程(大連)有限公司;丙烯酰胺葡聚糖凝膠(SephacryI S-200 HR) 北京天恩澤生物技術(shù)有限公司;Trizol試劑 Invitrigen公司;異丙基硫代半乳糖苷(isopropylβ-D-thiogalactoside,IPTG)、卡納霉素、氯化銫(均為分析純) 北京索萊寶科技有限公司。
1.2 儀器與設(shè)備
LightCycler 2.0實時熒光PCR儀 瑞士Roche公司;NanoPhotometer Pearl微量核酸蛋白測定儀 德國Implen公司;JEM-1200透射電鏡 日本電子公司;Infinity 3006凝膠成像系統(tǒng) 法國Vilber Lourmat公司;Lambda750紫外-可見近紅外分光光度計 美國PerkinElmer公司;BS-100A自動部分收集器 上海青浦滬西儀器廠。
1.3 方法
1.3.1 引物與探針
引物與探針由北京六合華大基因科技有限公司(以下簡稱華大基因)合成,相關(guān)信息見表1。
1.3.2 目的片段QβMulV和MulV的合成
根據(jù)ISO/T 15216-2 2012、GB/T 22287—2008《貝類中甲型肝炎病毒檢測方法 普通RT-PCR方法和實時熒光RT-PCR方法》、SN/T 2520—2010《貝類中A群輪狀病毒檢測方法 普通PCR和實時熒光PCR方法》、SN/T 2519—2010《貝類中星狀病毒檢測方法 普通PCR和實時熒光PCR方法》等國際標準、國家標準、行業(yè)標準分別規(guī)定的NoV(GI型和GII型)、HAV、RV、AstV檢測靶標,由華大基因合成同時含有上述4 種病毒檢測靶標RNA對應cDNA的串聯(lián)序列,命名為MulV。
參考GenBank數(shù)據(jù)庫中的Qβ噬菌體基因組序列(AB971354),由華大基因合成包含Qβ噬菌體成熟酶編碼基因、衣殼蛋白編碼基因、包裝位點序列、MulV的核酸片段,命名為QβMulV。
1.3.3 重組質(zhì)粒構(gòu)建與鑒定
將核酸片段MulV亞克隆至pGEM-T-Easy載體中,構(gòu)建體外轉(zhuǎn)錄用重組質(zhì)粒,酶切鑒定后送至華大基因測序。
參考同源重組克隆試劑盒的說明書,將核酸片段QβMulV亞克隆到pET-28a(+)載體中,構(gòu)建原核表達用重組質(zhì)粒,酶切鑒定后送至華大基因測序。
1.3.4 病毒樣顆粒的誘導表達
將原核表達用重組質(zhì)粒轉(zhuǎn)入E. coli BL21(DE3)感受態(tài)細胞,涂布于含卡那霉素的LB固體培養(yǎng)基上,37 ℃過夜培養(yǎng)后挑取單菌落,接種到5 mL LB液體培養(yǎng)基中37 ℃、180 r/min振蕩培養(yǎng)過夜,取2 mL菌液接種于200 mL LB液體培養(yǎng)基中,37 ℃、180 r/min振蕩培養(yǎng)至OD600nm約為0.6,加入終濃度為0.8 mmol/L IPTG誘導表達12 h,離心收集菌液,進行十二烷基硫酸鈉-聚丙稀酰胺凝膠電泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)分析。
1.3.5 病毒樣顆粒的純化
將1.3.4節(jié)收集的菌液采用超聲波破碎,4 ℃、11 000 r/min離心20 min收集上清后分別加入終濃度為8 U/mL的DNase I和12 μg/mL的RNase A消化,用0.45 μm濾膜過濾后進行氯化銫密度梯度離心,4 ℃、80 000 r/min離心5 h,小心吸取含VLPs的目標層液體,通過SephacryI S-200 HR層析柱進一步純化,用自動樣品收集器每10 min收集一管樣品。將各收集管樣品測定在260 nm和280 nm波長處的吸光度,并進行SDS-PAGE分析。獲得同時含有4 種食源性病毒檢測靶標的多聯(lián)裝甲RNA,命名為AR-MulV。
1.3.6 電鏡觀察
將AR-MulV用2%磷鎢酸負染,在JEM-1200型透射電子顯微鏡下觀察VLPs形態(tài)。
1.3.7 AR-MulV中殘留質(zhì)粒的檢測
以純化后的AR-MulV為模板,用實時熒光定量PCR法檢測其中是否含有殘留重組質(zhì)粒;同時設(shè)置pETQβMulV為陽性對照,無酶ddH2O為空白對照;每個樣品設(shè)置3 個平行。檢測用引物與探針參見表1,反應體系見表2,循環(huán)參數(shù):95 ℃預變性10 s;95 ℃變性5 s,60 ℃退火延伸20 s,40 個循環(huán)。
1.3.8 AR-MulV的初步定值1.3.8.1 cRNA制備
用限制性內(nèi)切酶線性化體外轉(zhuǎn)錄用重組質(zhì)粒,用T7 RiboMAXTMExpress Large Scale RNA Production System試劑盒進行體外轉(zhuǎn)錄;體系如下:RioMAXTMExpress T7 2 μL,Buffer 10 μL,Linear DNA Template 8 μL,Enzyme Mix T7 Express 2 μL;37 ℃溫育30 min后,加入1 μL的RQ1 RNase-Free DNase,37 ℃溫育15 min,除去DNA模板。用Trizol法提取cRNA。
1.3.8.2 標準曲線構(gòu)建
使用微量核酸蛋白測定儀測定cRNA的濃度。計算拷貝數(shù)按照:拷貝數(shù)/(copies/μL)=(6.02×1023拷貝/mol×RNA含量/(ng/μL)×10-9)/(片段長度/bp×340),將cRNA進行10 倍梯度稀釋,用對應的引物與探針分別對4 種食源性病毒檢測靶標進行實時熒光定量RT-PCR分析,每個樣品設(shè)置3 個平行,體系見表3。循環(huán)參數(shù)為42 ℃反轉(zhuǎn)錄5 min,95 ℃變性10 s;95 ℃變性5 s,60 ℃退火延伸20 s,40 個循環(huán)。根據(jù)所測得的Ct值與cRNA各梯度稀釋拷貝數(shù)的對數(shù)繪制標準曲線。
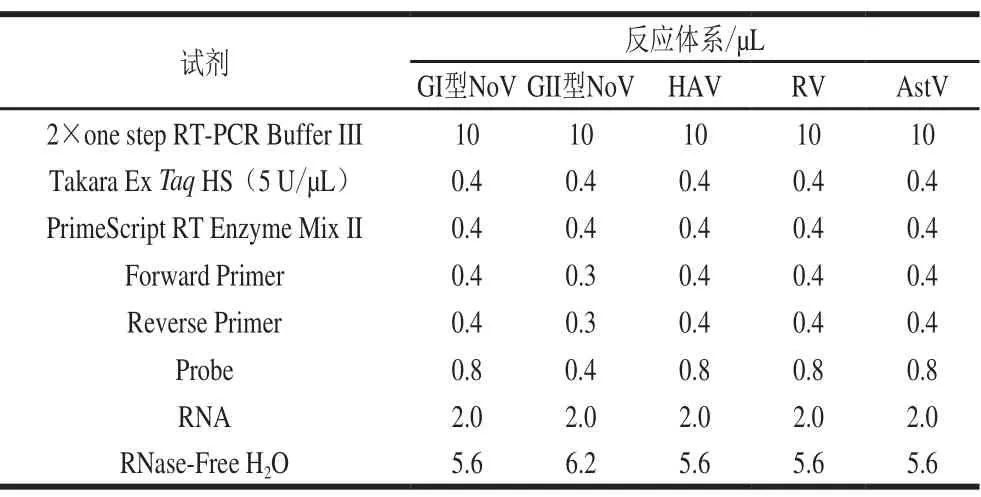
表 3 4 種食源性病毒的實時熒光定量RT-PCR體系Table 3 Real-time RT-PCR system for detection of four foodborne viruses
1.3.8.3 初步定值與不確定度分析
將純化后的AR-MulV用無酶磷酸緩沖鹽溶液(phosphate buffer saline,PBS)(pH 7.2)適度稀釋,分裝到1.5 mL無酶離心管中,參考JJF 1006—1994《一級標準物質(zhì)技術(shù)規(guī)范》的要求,隨機選取15 管AR-MulV,采用1.3.8.2節(jié)中的實時熒光定量RT-PCR方法分別測定各食源性病毒的Ct值,根據(jù)標準曲線,得到15 管AR-MulV中4 種食源性病毒5 個檢測靶標拷貝數(shù),參考JJF 1006—1994中格布拉斯準則剔除可疑數(shù)據(jù)后,計算平均值,得到AR-MulV中各食源性病毒檢測靶標初步定值結(jié)果。不確定度分析參考JJF 1059—1999《測量不確定度表示指南》,采用A類不確定度評定方法,按照以下公式進行計算:
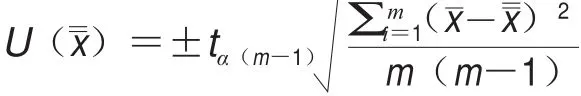
式中:tα(m-1)為顯著性水平α、自由度為m-1的t值;為單次測量的平均值;為測量的總平均值;m為測量次數(shù)。
2 結(jié)果與分析
2.1 重組質(zhì)粒的構(gòu)建
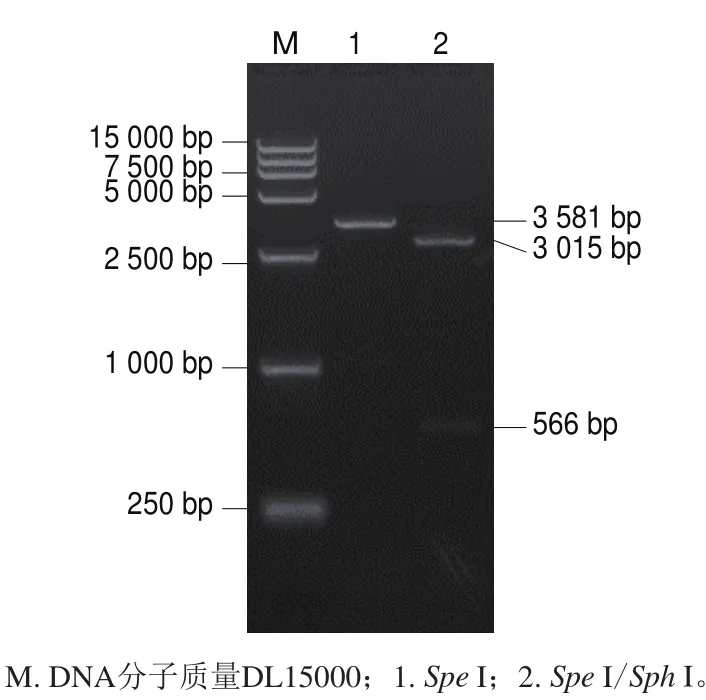
圖 1 pGEM-MulV的酶切鑒定電泳圖Fig. 1 Enzyme digestion of pGEM-MulV
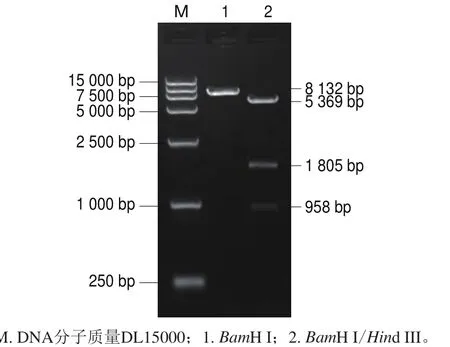
圖 2 pET-QβMulV的酶切鑒定電泳圖Fig. 2 Enzyme digestion of pET-QβMulV
分別用限制性內(nèi)切酶Spe I單酶切和Spel I/Sph I雙酶切鑒定體外轉(zhuǎn)錄用重組質(zhì)粒,大小與預期一致,電泳結(jié)果如圖1所示,測序結(jié)果正確,證實重組質(zhì)粒構(gòu)建成功,命名為pGEM-MulV。分別用限制性內(nèi)切酶BamH I單酶切和BamH I/Hind III雙酶切鑒定原核表達用重組質(zhì)粒,大小與預期一致,電泳結(jié)果如圖2所示,測序結(jié)果正確,證實重組質(zhì)粒構(gòu)建成功,命名為pET-QβMulV。
2.2 pET-QβMulV在大腸桿菌中的誘導表達
pET-QβMulV轉(zhuǎn)化入大腸桿菌BL21(DE3)后,經(jīng)IPTG誘導表達,用SDS-PAGE檢測,在約14.1 kDa處出現(xiàn)一條明顯的重組蛋白表達條帶(圖3),大小與預期一致,表明Qβ噬菌體衣殼蛋白成功表達。
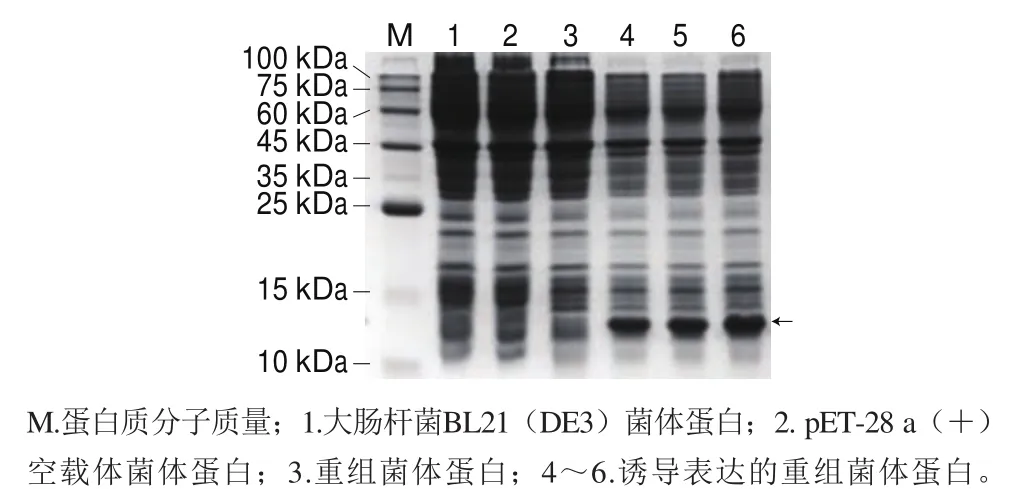
圖 3 表達產(chǎn)物的SDS-PAGE分析Fig. 3 SDS-PAGE analysis of expression product
2.3 樣品的純化
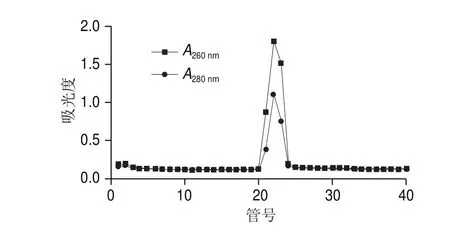
圖 4 丙烯葡聚糖凝膠層析純化不同收集管樣品吸光度分析結(jié)果Fig. 4 Absorbance of different fraction after purification by Sephacryl S-200 HR gel exclusion chromatography

圖 5 吸收峰附近樣品SDS-PAGE結(jié)果Fig. 5 SDS-PAGE analysis of samples around absorbent peak
如圖4所示,第21管在260 nm和280 nm波長處均有最大吸光度,說明該管中核酸(即靶標RNA)與蛋白(即VLPs)含量均最高,即該管中AR-MulV含量最高。第19~24管樣品的SDS-PAGE分析結(jié)果見圖5,可見第21、22管樣品在14.1 kDa處均有條帶且濃度高無雜帶,說明純化效果好。本研究選取第21管樣品用無酶PBS(pH 7.2)適度稀釋后進行后續(xù)實驗。
2.4 電鏡觀察結(jié)果
在電鏡下觀察純化后的AR-MulV,可見大量結(jié)構(gòu)完整、大小均一的病毒樣顆粒,直徑約25 nm,與預期大小一致,結(jié)果如圖6所示。
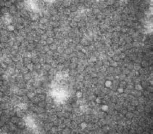
圖 6 AR-MulV電鏡照片F(xiàn)ig. 6 Electron microscopy image of AR-MulV
2.5 AR-MulV中殘留質(zhì)粒的檢測結(jié)果
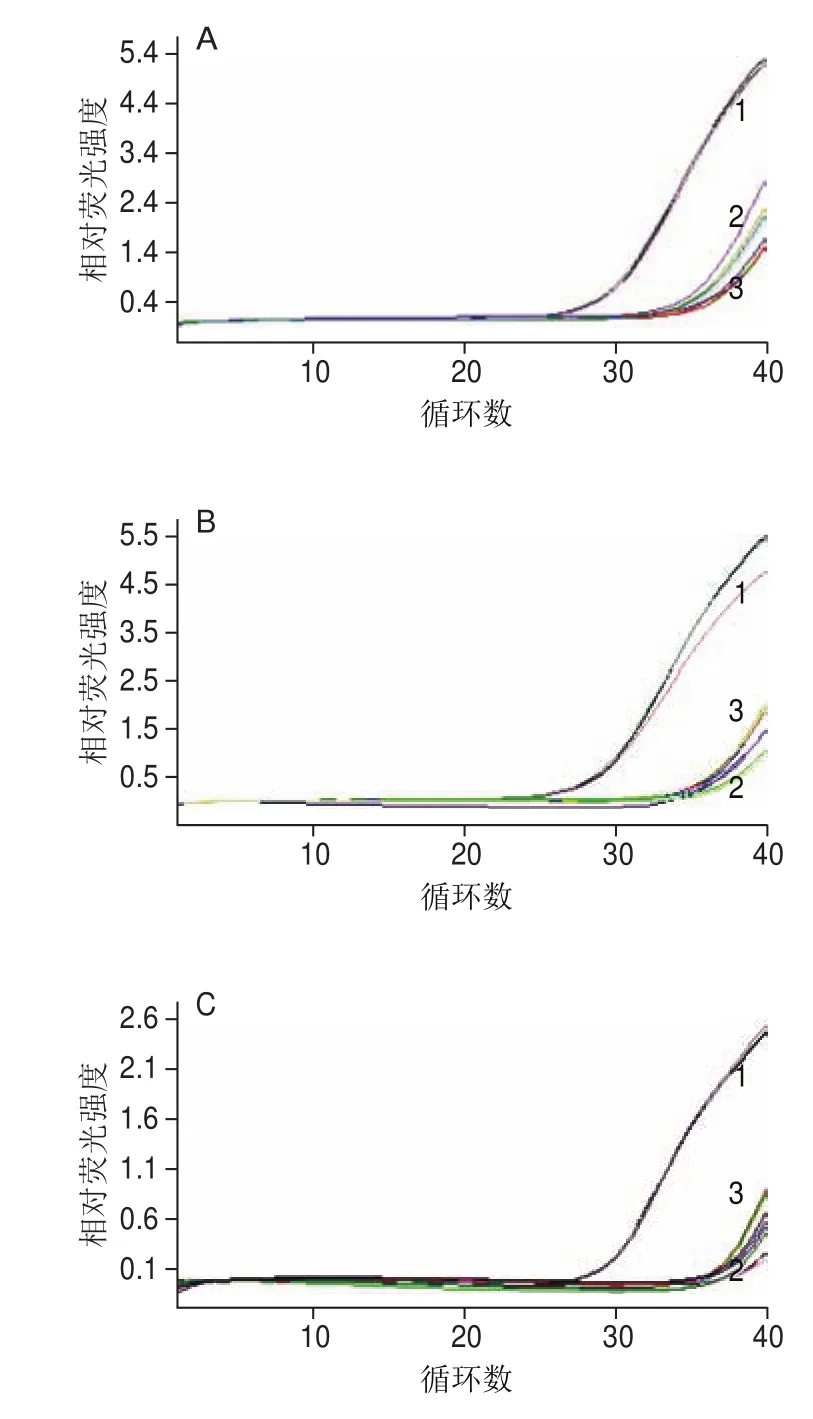
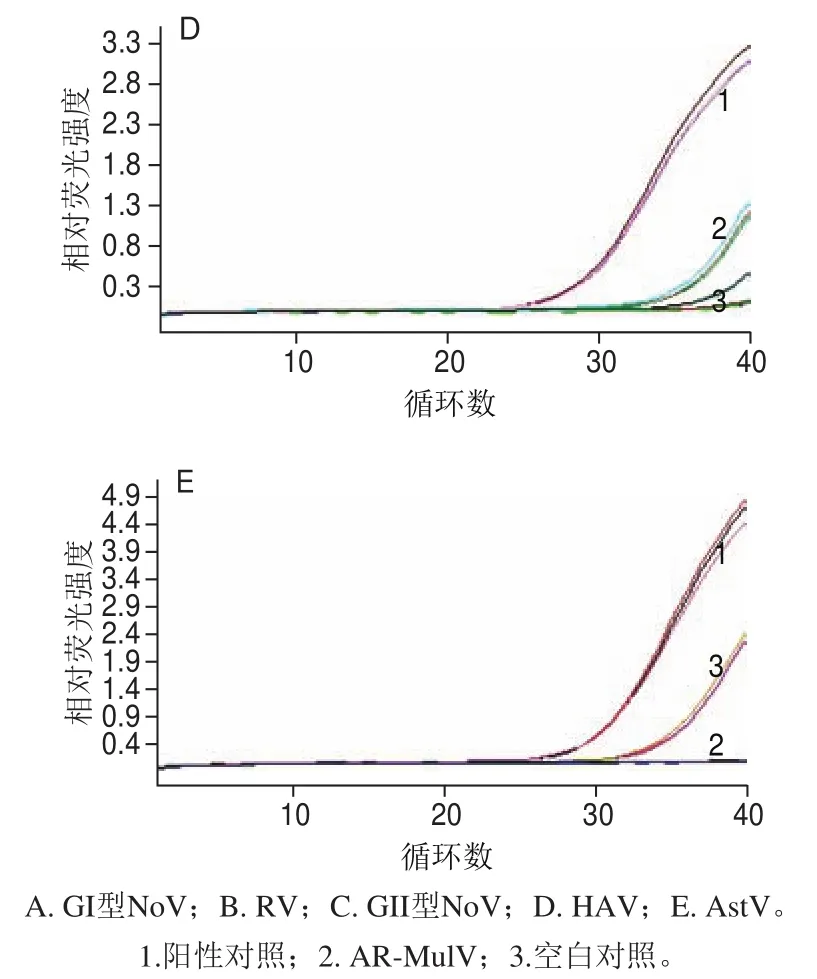
圖 7 AR-MulV殘留質(zhì)粒實時熒光定量PCR檢測結(jié)果Fig. 7 Real-time PCR for residual plasmid DNA detection in the AR-MulV
如圖7所示,以AR-MulV為模板,分別用4 種食源性病毒的5 個檢測靶標對應的引物和探針進行檢測的實驗組和空白對照組,在陰性判定Ct值內(nèi)均未出現(xiàn)特異性擴增;以pET-QβMulV為陽性對照的實驗組每對引物與探針均出現(xiàn)明顯的特異性擴增,且Ct值均在陽性判定值內(nèi),說明純化的AR-MulV中無重組質(zhì)粒殘留,純度良好。
2.6 AR-MulV初步定值
2.6.1 cRNA標準曲線的構(gòu)建
以10 倍稀釋梯度的cRNA為模板,利用4 種食源性病毒的引物與探針,分別進行實時熒光定量RT-PCR檢測。以cRNA各稀釋梯度的對數(shù)為橫坐標,以Ct值為縱坐標建立實時熒光定量RT-PCR的標準曲線,其線性方程見表4。

表 4 4 種食源性病毒的cRNA標準曲線Table 4 Calibration curves for cRNA of four foodborne virus
2.6.2 AR-MulV的初步定值與不確定度分析
用對應的實時熒光定量RT-PCR方法檢測15 管AR-MulV,計算獲得AR-MulV中該食源性病毒靶標核酸拷貝數(shù),同時計算A類不確定度。定值結(jié)果見表5,AR-MulV中GI型NoV、GII型NoV、HAV、RV和AstV檢測靶標RNA的含量分別為(1.24±0.2)×107、(2.54±0.6)×107、(2.24±0.3)×107、(2.96±0.5)×107copies/μL和(3.19±0.4)×107copies/μL。
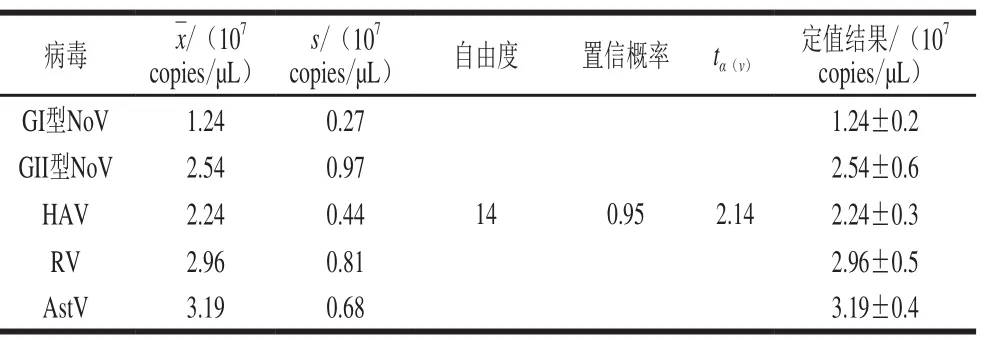
表 5 AR-MulV中4 種食源性病毒靶標RNA初步定值結(jié)果Table 5 Quantification of AR-MulV for 4 foodborne viruses
3 討 論
目前食源性病毒檢測標準中的陽性質(zhì)控樣品主要有致弱的活病毒或cDNA/質(zhì)粒DNA兩大類,前者需要進行病毒培養(yǎng),存在一定生物安全隱患,并且需使用與目標病毒不同的引物、探針,操作繁瑣;后者無法監(jiān)控病毒裂解、核酸提取、反轉(zhuǎn)錄等多個關(guān)鍵環(huán)節(jié)。此外,也有將體外轉(zhuǎn)錄cRNA作為陽性質(zhì)控樣品[8-10,15],雖然可以評價反轉(zhuǎn)錄的過程,但存在易降解的短板。利用裝甲RNA技術(shù)制備的陽性質(zhì)控樣品在最大程度上模擬病毒粒子,可在檢測過程中添加到實際樣本或空白樣本中,對目標病毒的裂解、RNA提取、檢測等關(guān)鍵環(huán)節(jié)進行全程評價,無生物安全隱患,穩(wěn)定性高。對于某些高變異性的RNA病毒,在確定檢測靶序列的前提下,可在28 d內(nèi)構(gòu)建、制備、純化出新的裝甲RNA[16],在自身有合成基因、測序條件的實驗室,不考慮定值,只制備定性陽性質(zhì)控樣品,周期可縮短至20 d;有研究表明,裝甲RNA的穩(wěn)定性很好,在-20 ℃條件下至少可保存120~200 d[14,25]、-80 ℃條件下至少可保存300 d[14],這種較短的制備周期與優(yōu)良的穩(wěn)定性也是裝甲RNA的重要優(yōu)勢[16]。裝甲RNA技術(shù)完全可彌補致弱的活病毒、cDNA/質(zhì)粒DNA和cRNA作為陽性質(zhì)控樣品的不足[17-18],因此該技術(shù)已成為RNA病毒核酸檢測陽性質(zhì)控樣品研發(fā)的重要發(fā)展方向[18-21],尤其對于烈性傳染性病毒、新發(fā)病毒和無法進行體外培養(yǎng)病毒而言有著無可替代的優(yōu)勢,現(xiàn)已成功制備人體免疫缺陷病毒[13]、猴免疫缺陷病毒[22]、冠狀病毒[23]、口蹄疫病毒[24]、寨卡病毒[16]、腸道病毒[12]等重要病原的裝甲RNA。
本研究在純化了AR-MulV后,為明確其所含各食源性病毒檢測靶標RNA的拷貝數(shù),使之今后能更好地用于定量評價檢測方法、儀器設(shè)備靈敏度等領(lǐng)域,探索性地對其進行初步定值研究。目前對于裝甲RNA的定值主要有3 種方式:1)使用線性化質(zhì)粒DNA為模板構(gòu)建標準曲線[22];2)使用體外轉(zhuǎn)錄cRNA為模板構(gòu)建標準曲線[25];3)以制備的裝甲RNA提取的RNA為模板構(gòu)建標準曲線[26-27]。有研究結(jié)果表明,體外轉(zhuǎn)錄的cRNA為標準品更接近樣本RNA的真實拷貝數(shù)[28],所以本研究以體外轉(zhuǎn)錄的cRNA為模板構(gòu)建標準曲線對制備的AR-MulV進行初步定值。結(jié)果表明,即使經(jīng)過稀釋,AR-MulV所含的4 種食源性病毒的檢測靶標RNA均可達到107copies/μL。在技術(shù)成熟的實驗室,理論上講,100 mL的誘導菌液可制備10 L以上的107copies/μL高拷貝數(shù)的樣品,產(chǎn)量大。后期對其進行聯(lián)合定值,明確其穩(wěn)定性、均勻性等重要特性后,將有望申報有證標準參考樣品。
NoV、HAV、RV、AstV這4 種食源性病毒已成為質(zhì)檢、疾控、出入境等機構(gòu)的重要檢測項目,本研究基于目前通行的國際標準、國家標準、行業(yè)標準中涉及的食源性病毒檢測靶標,制備多聯(lián)裝甲RNA,與分別制備的方法相比,節(jié)省時間與研究精力。此外,為滿足食品安全與公共衛(wèi)生安全等領(lǐng)域的需要,多重實時熒光定量RT-PCR成為快速篩查食源性病毒的重要手段,目前國內(nèi)外學者已成功建立了NoV、HAV、RV和AstV[1],GI型和GII型NoV[29],NoV、RV、HAV和柯薩奇病毒[30],GI型、GII型NoV和HAV[31]等多重實時熒光定量RT-PCR檢測方法,能夠快速、特異、靈敏地同時檢測多種食源性病毒。在應用上述多重檢測方法時,如果對涉及到的每種食源性病毒均設(shè)置單一的陽性質(zhì)控樣品,則需同時對多種相應的質(zhì)控樣品分別添加、提取、擴增等,操作繁瑣,難以滿足應對突發(fā)性食品安全與公共衛(wèi)生事件對檢測時效性的要求。針對上述需求,可基于本研究開發(fā)的多聯(lián)裝甲RNA研發(fā)平臺,根據(jù)多重實時熒光定量RT-PCR方法對應的擴增靶標序列,快速制備多聯(lián)裝甲RNA,滿足多種病毒的快速高通量檢測對陽性質(zhì)控樣品的需求。多重實時熒光定量RT-PCR檢測方法與多聯(lián)裝甲RNA聯(lián)合使用,可縮短樣品檢測時間,提高檢測結(jié)果的可靠性。
綜上所述,本研究基于Qβ噬菌體裝甲RNA技術(shù),成功研制出同時包含NoV、HAV、RV和AstV 4 種食源性病毒檢測靶標的多聯(lián)裝甲RNA,無殘留DNA,RNA拷貝數(shù)高,后期經(jīng)穩(wěn)定性、均勻性評價后,可作為多種病毒分子檢測的陽性質(zhì)控樣品供質(zhì)檢、科研人員選擇使用,為食源性病毒標準化檢測提供安全的標準參考樣品提供參考依據(jù),也為多重實時熒光定量RT-PCR配套使用的多聯(lián)陽性質(zhì)控樣品的制備提供了新思路。
猜你喜歡
中國設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:36
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:34
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:50
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:48
