低溫等離子體制備低碳醇合成用KNiMo基催化劑及其結構性能表征
2019-06-03 02:32:12劉曉展曾春新房克功
燃料化學學報 2019年5期
關鍵詞:催化劑
李 瑩, 趙 璐, 劉曉展, 曾春新, 房克功
(1. 中國科學院山西煤炭化學研究所 煤轉化國家重點實驗室, 山西 太原 030001;2. 中國科學院大學, 北京 100049)
由于低碳混合醇的潛在應用價值,合成氣制低碳醇(HAS)研究受到科研工作者的廣泛關注[1,2]。然而,因催化劑穩定性較差以及低碳醇時空產率較低等原因,現有技術仍然不能滿足工業應用[3]。目前,HAS催化劑主要有以下幾類:貴金屬基(Rh基)催化劑[4,5]、Cu改性費托合成(Co-、Fe-、Ni-)基催化劑[6-10]、改性CuZnAl基(Cu基)甲醇合成催化劑[11,12]和MoS2基催化劑[13-15]。盡管貴金屬(Rh基)催化劑對乙醇具有高選擇性,但貴金屬的稀缺性和高昂的價格限制了其在商業中的應用。具有較低成本的Cu改性Co-、Fe-、Ni-基費托合成催化劑在溫和條件下表現出較高的反應活性,但該類催化劑顯現出較高的烴類選擇性和較低的熱穩定性。改性CuZnAl基(Cu基)催化劑往往產出甲醇和丁醇,對C2+醇的選擇性相對較低。
MoS2基催化劑是一類具有前景的非貴金屬催化劑。由于反應過程中不易積炭、抗硫中毒能力強并且有著相對高的醇類選擇性,因此,得到廣泛研究。但MoS2基催化劑也有缺點:如反應活性比金屬催化劑低、需要更高的反應壓力和溫度才能達到較高的時空產率、難以避免將含硫雜質引入最終產品等。大量研究已表明,未改性的MoS2在CO加氫反應中主要產物為烴類,而堿金屬K助劑的引入可以促進醇的形成[16,17]。基于廣泛共識的CO插入機理[18],引入堿金屬K可以提高催化劑對CO的非解離吸附能力、抑制CHx物種的加氫,進而減少烴的生成并顯著增加醇類選擇性[19]。此外,已有大量文獻報道,在K改性MoS2中添加Ni作為助劑能夠明顯提高催化劑的C2+醇選擇性和總醇時空產率。這種KNiMo基催化劑具有優異HAS催化性能主要歸因于Ni的引入有效改善了活性相的分散度和催化CO插入及碳鏈增長的能力[15,20]。
研究發現,對于MoS2基催化劑,反應分子化學吸附在其邊、角位而不是MoS2片層的基面位,位于邊、角的配位不飽和活性位通常作為HAS的催化中心[21,22],因而MoS2基催化劑的催化性能通常取決于邊、角位與基面位的相對比例。該比例與MoS2片層的長度、層數、剝離程度等因素關聯,而這些因素又與其合成方法密切相關。傳統MoS2基催化劑的合成過程包含熱處理環節,如采用共沉淀法、浸漬法等得到鉬基前體后需經高溫焙燒、熱硫化處理。但是上述制備方法常常導致Mo基活性相的團聚、燒結,這些均不利于CO加氫合成低碳醇[23]。因此,為了提高催化劑的催化性能,探究適宜的新制備方法用以合成尺度小、片層數低的MoS2基催化劑至關重要。
近年來,低溫等離子體技術在催化劑合成領域被廣泛研究[24,25]。與傳統方法相比,低溫等離子體技術具有能耗低、合成時間短和制備的催化劑顆粒粒徑小等優點[26]。Jiang等[27]使用低溫等離子體法制備了尖晶石相的LiMn2O4。由于其粒徑小且分布集中而具有優異的電荷傳導性能,表現出良好的鋰離子電池充放電性能和循環穩定性。Li等[28]開發了一種低溫等離子體制備Pd/Al2O3催化劑的方法,改善了Pd顆粒的分散,獲得了優異的乙炔轉化活性和高的乙烯選擇性。張旭等[29]通過浸漬法合成了Ni/SiO2催化劑并采用低溫等離子體技術對其進行了改性,明顯增加了催化劑活性相分散度及對反應物的吸附強度,CO2甲烷化反應性能顯著提高。Tan等[30]使用低溫等離子體法去除ZnAl-水滑石上負載的Au納米顆粒的保護配體,獲得了最佳的催化性能,表明該法不僅可以解離水滑石中的層間離子,而且還可以打斷Au與保護劑之間的化學鍵。Wang等[31]報道了一種由低溫等離子體制備的NiMgSBA-15催化劑,發現改善了載體與Ni的相互作用并減小了Ni的顆粒粒徑,從而提高了催化性能。
本研究采用低溫等離子體法合成出了系列低碳醇合成用KNiMo基催化劑。通過XRD、BET、TEM、HAADF-STEM、H2-TPD、CO-TPD、原位CO吸附DRIFTS等技術手段對所合成催化劑進行了表征分析,并與傳統熱法合成的KNiMo基催化劑進行了物化性質、HAS催化反應性能等的對比研究。
1 實驗部分
1.1 催化劑的制備
采用溶膠凝膠法合成KNiMo基催化劑前體(以Mn-Al復合物為基底)。所用試劑原料均為化學分析純。前體制備過程為:先將一定量的硝酸錳、硝酸鋁、檸檬酸(CA)用去離子水溶解制得水溶液(Mn∶Al∶CA=5∶3∶10)[32,33]。向上述溶液中滴入一定量的聚乙二醇(PEG400),攪拌1.5 h,得到溶液A。將硝酸鉀、硝酸鎳和鉬酸銨在室溫下溶解,其中,K∶Mo∶Ni =3∶5∶1.6。以物質的量比CA/Mo=2.5向上述溶液中引入CA后65 ℃攪拌2 h,得到澄清溶液B。在劇烈攪拌下,將溶液A和溶液B均勻混合并于80 ℃攪拌3 h得到凝膠。120 ℃干燥上述凝膠即得到KNiMo基催化劑前體。
將上述前體分成四部分:第一部分直接用含H2S等離子體處理(標記為KNiMo-DPS);第二部分在500 ℃空氣氣氛下焙燒3 h后利用H2S等離子體硫化(標記為KNiMo-CPS);第三部分在H2S/H2氣氛中500 ℃下直接處理3 h(標記為KNiMo-DTS);第四部分在500 ℃空氣氣氛下焙燒3 h后再于H2S/H2氣氛中進行500 ℃熱硫化處理(標記為KNiMo-CTS)。圖1(a)為介質阻擋放電(DBD)等離子體反應器的結構示意圖。反應氣氛為流量60 mL/min的60%H2S/H2,穩定狀態下的總輸入功率約為30 W,合成時間約為20 min。通過紅外成像儀確定實際等離子體放電合成條件下反應器床層的溫度分布。如圖1(b)所示,反應器床層的平均溫度約為95.1 ℃,因此,可以忽略低溫等離子體合成過程中的熱效應。
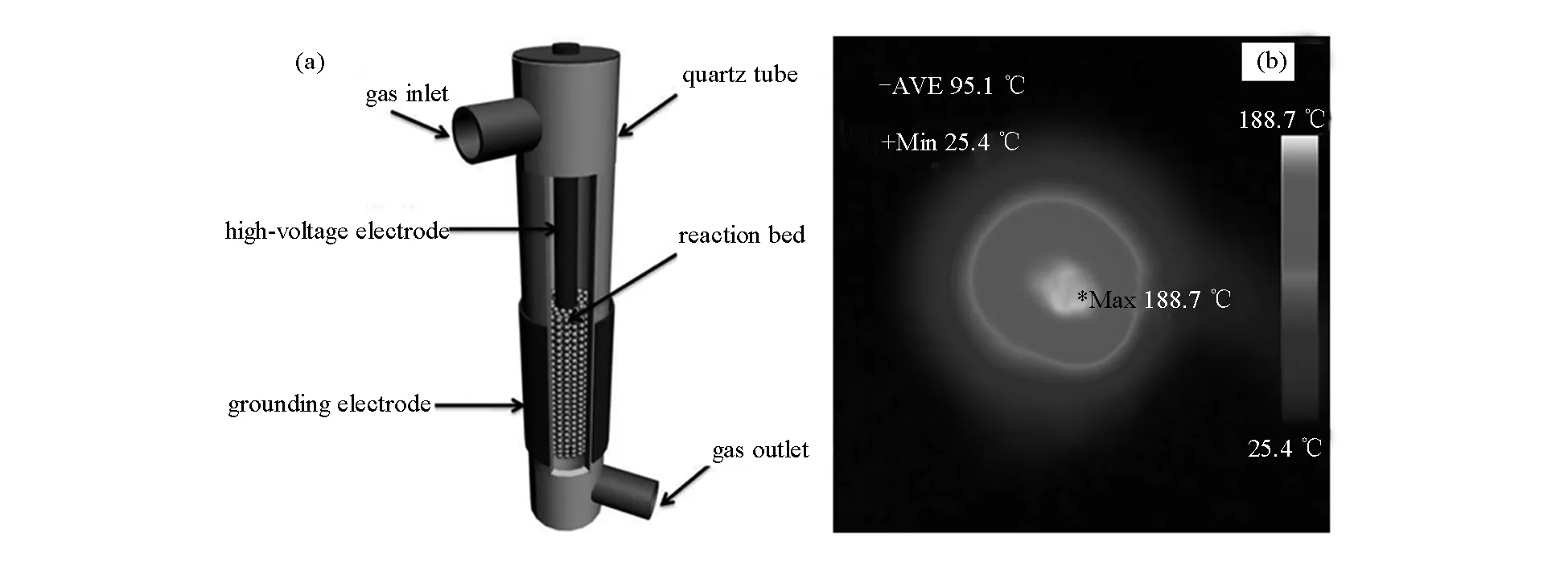
圖 1 (a) 硫化用介質阻擋放電(DBD)等離子體反應器結構示意和(b) DBD反應器放電時床層溫度分布
1.2 催化劑的表征
X射線衍射(XRD)測試在Rigaku D/Max-RA衍射儀上進行。以CuKα為輻射源,其中,管電壓40 kV。采用Micromeritics ASAP-2000進行催化劑比表面積的測定。透射電子顯微鏡(TEM)照片和高角環形暗場(HAADF-STEM)照片通過Tecnai G2 F20 S-Twin顯微鏡在200 kV下采集。H2程序升溫脫附(H2-TPD)、 CO程序升溫脫附(CO-TPD)在常壓石英反應器中進行。原位CO吸附DRIFTS在Bruker Vertex 80v FT-IR光譜儀上進行檢測。作者已在前期研究中詳細闡述了以上表征過程,請參見文獻[34,35]。
1.3 CO加氫合成低碳醇反應性能的評價
將催化劑(40-60目,1.5 mL)與1.5 mL石英砂均勻混合,裝入內徑10 mm的不銹鋼固定床反應器中。反應條件為反應溫度350 ℃、反應壓力5.0 MPa、體積空速5000 h-1、原料氣氫碳比(H2/CO)= 2。合成氣經質量流量計控制進入反應器,反應經24 h后達到穩態進行取樣分析。通過熱阱(120 ℃) 和冷阱(0 ℃)收集液相產物。采用氣相色譜完成氣、液相產物分析。H2、CO通過TDX-01分子篩色譜柱分析,Ar為載氣,TCD檢測;CO、CH4及CO2采用TDX-01分子篩色譜柱分析,He為載氣,TCD檢測;C1-10烴產物(烷烴、烯烴等)通過Al2O3色譜柱分析,Ar為載氣,FID檢測;液相通過兩根Porapak-Q色譜柱分析,分別以TCD和FID檢測。其中,TCD檢測甲醇和水,FID檢測C1-5OH (正構和異構醇)。上述分析中以CH4關聯氣相產物,以甲醇關聯液相產物進行歸一化計算。
2 結果與討論
2.1 XRD分析
圖2為四種方法合成的KNiMo基催化劑的XRD譜圖。由圖2可知,在29.7°、34.4°、49.0°和60.8°出現了四個特征衍射峰,分別歸屬于NiS的(100)、(101)、(131)和(103)晶面的特征衍射峰。
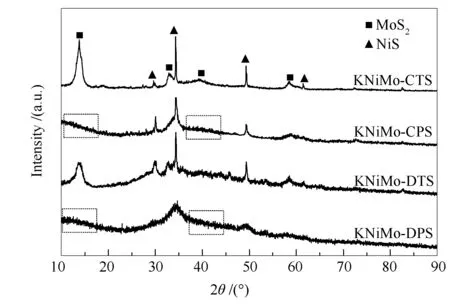
圖 2 不同方法合成的KNiMo基催化劑的XRD譜圖
14°的衍射峰對應于MoS2(002)晶面的特征衍射。對比低溫等離子體法制備的KNiMo基催化劑(KNiMo-DPS和KNiMo-CPS)與熱法制備的催化劑(KNiMo-DTS和KNiMo-CTS)的XRD譜圖發現,前者未觀察到該(002)晶面的特征衍射峰。表明沿c軸方向的MoS2片層堆疊不易發生,在等離子體制備的KNiMo基催化劑中層狀結構的片層層數小于5[36]。這與后文TEM照片觀察到的結果一致。此外,KNiMo-DPS和KNiMo-CPS催化劑的XRD衍射峰峰形寬化且峰強度低,表明其活性相顆粒粒徑小,分散度高。由表1可知,由Scherrer方程計算得到的KNiMo-DTS和KNiMo-CTS催化劑的平均顆粒粒徑分別為35.8和29.6 nm,而KNiMo-CPS的顆粒粒徑為8.7 nm。KNiMo-DPS的顆粒粒徑相對更小,約7.1 nm。活性相顆粒粒徑的減小可以增加不飽和活性位(邊、角位)的數量,同時也有效改善了分散度,但經熱法處理會導致活性相的團聚長大,進而導致催化性能的降低。與此同時,在KNiMo-DPS和KNiMo-CPS催化劑上未觀察到MoS2(103)晶面的特征衍射峰,該(103)晶面衍射峰的缺失表明等離子體制備方法有利于形成少層、無序結構[37],進而提高了KNiMo基催化劑活性位數量。
由表1可知,KNiMo-DPS具有最大的比表面積,為51 m2/g,而KNiMo-CTS的比表面積最小,僅為23 m2/g。這一結果顯示低溫等離子體方法可以增大樣品的比表面積,從而有效增加活性相的分散度。與傳統熱法相比,低溫等離子體法合成所需時間短、溫度低,這可以避免活性相顆粒的聚集長大,并增大了催化劑的比表面積,這些均有利于催化反應的進行。
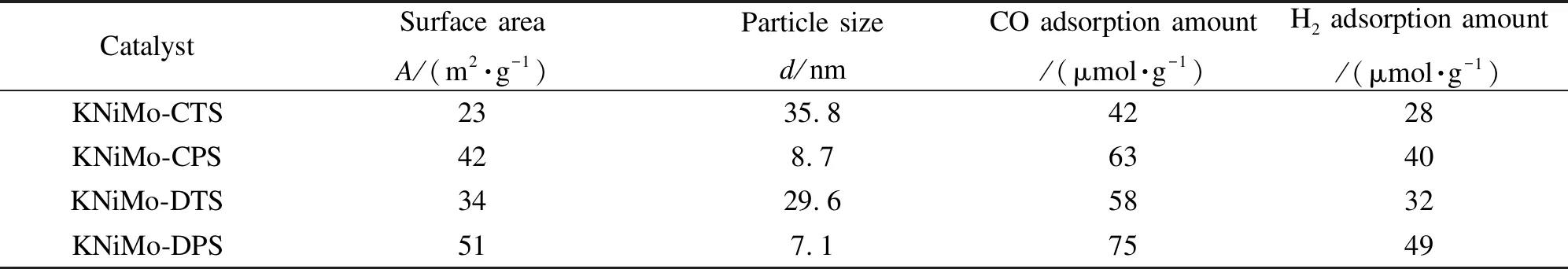
表 1 不同方法合成的KNiMo基催化劑的物化性質
2.2 TEM分析
采用TEM分析進一步觀測不同方法合成的KNiMo基催化劑的形貌和顆粒粒徑。不同催化劑的TEM照片見圖3。

圖 3 不同方法合成的KNiMo基催化劑的TEM照片
圖3(a)和(c)示出了KNiMo-CTS和KNiMo-DTS催化劑顆粒具有多層結構。與熱法合成催化劑相比,低溫等離子體方法制備的KNiMo-DPS和KNiMo-CPS催化劑顯示顆粒粒徑約7-10 nm。如圖3(b)和(d)所示,顆粒粒徑明顯小于KNiMo-DTS和KNiMo-CTS催化劑(長度30-50 nm),與XRD表征相一致。圖3中TEM照片顯示低溫等離子體法可以得到粒徑更小的顆粒。TEM照片中也觀察到KNiMo-DPS和KNiMo-CPS催化劑中的活性相顆粒高度分散且分布更加無序。此外,KNiMo-DTS和KNiMo-CTS沿c軸堆疊的MoS2片層層數(15-25層)也明顯高于KNiMo-DPS和KNiMo-CPS催化劑(1-3層)。因此,通過低溫等離子體法合成的KNiMo基催化劑明顯提高了邊、角位與基面位的比例,為催化反應提供了更多的邊、角位的催化活性中心,這些改進均利于催化反應的進行。該TEM分析結果也與XRD分析結果一致。圖4為KNiMo-DPS催化劑的HAADF-STEM照片。由圖4可知,K、Ni、Mo和S元素的EDS照片證實了各元素均勻分散并且相伴共存。進一步說明低溫等離子體法可制備出具有良好結構穩定性、均勻分散的KNiMo基催化劑。
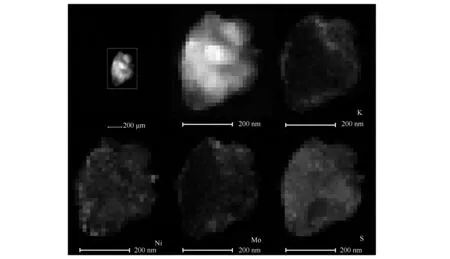
圖 4 KNiMo-DPS催化劑的HAADF-STEM-EDS照片
2.3 H2-TPD分析
圖5為不同方法所制備KNiMo基催化劑的H2-TPD譜圖。在四組不同樣品中均觀察到位于500-750 ℃的H2脫附峰。該脫附峰可歸屬于KNiMo基催化劑上H物種的強化學吸附,這與文獻報道的結果相一致[38]。由表1可知,研究中采用高斯函數擬合計算脫附氫量。觀測到的氫脫附量由高到低的順序依次為KNiMo-DPS > KNiMo-CPS > KNiMo-DTS > KNiMo-CTS,其中,KNiMo-DPS催化劑的氫脫附量最高,為49 μmol/g,近乎是KNiMo-CTS的兩倍(28 μmol/g),這意味著KNiMo-DPS催化劑具有更多的氫吸附活性位點,這也表明了通過低溫等離子體法可以有效改善KNiMo基催化劑的氫活化能力,從而提高CO加氫反應活性。
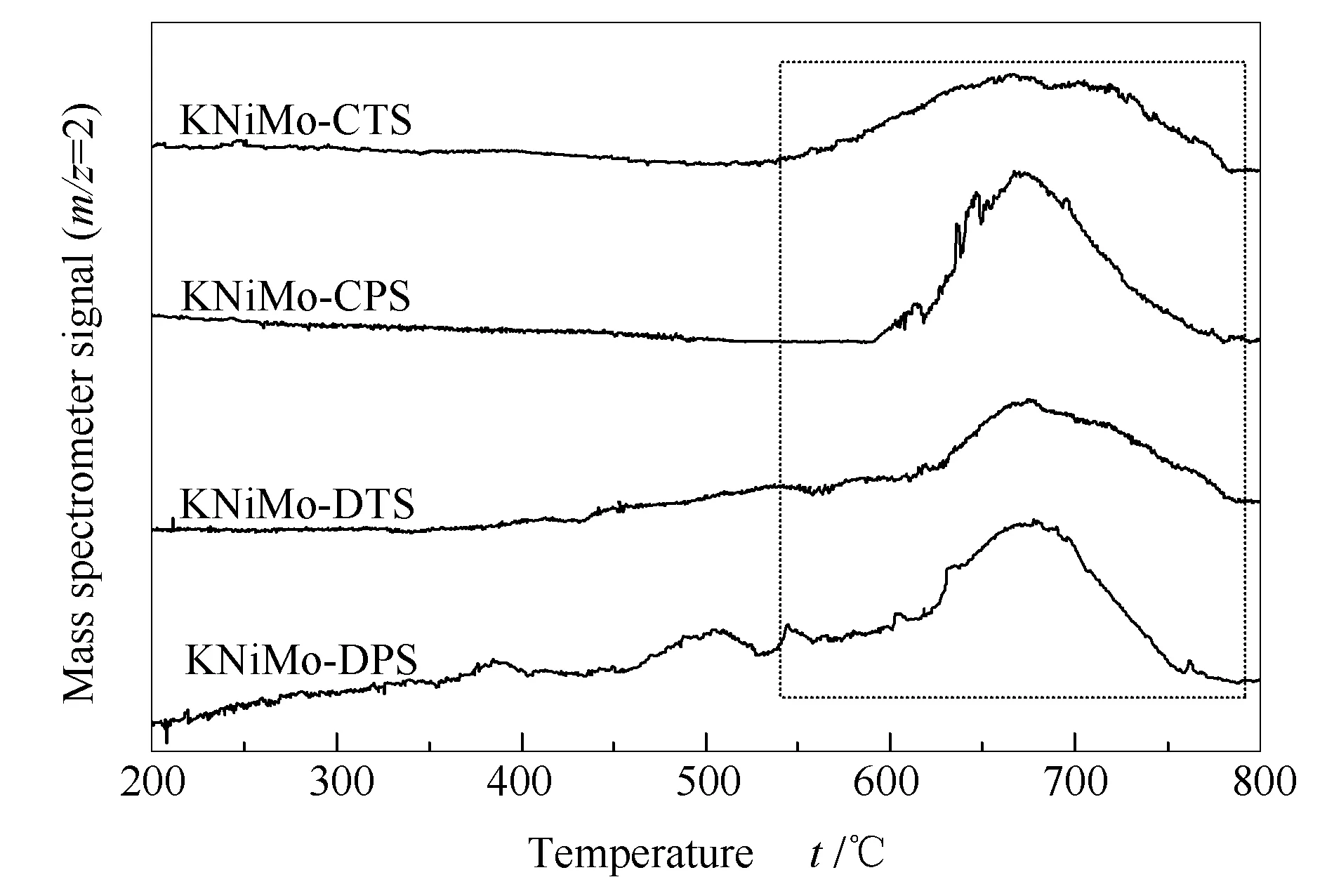
圖 5 不同方法所制備KNiMo基催化劑的H2-TPD譜圖
2.4 CO-TPD分析
不同KNiMo基催化劑的CO-TPD譜圖見圖6。對于各個催化劑均觀察到CO脫附峰出現在300-500 ℃。根據文獻報道,低碳醇合成用催化劑表面吸附的非解離CO物種主要參與了成醇過程中CO插入反應[39]。通過熱法合成的KNiMo-DTS和KNiMo-CTS催化劑在350-390 ℃出現寬化,強度較弱的CO脫附峰對應于催化劑上CO的弱吸附位點,表明KNiMo-DTS和KNiMo-CTS催化劑具有較低的CO非解離吸附能力。而通過低溫等離子體法合成的KNiMo基催化劑(KNiMo-DPS和KNiMo-CPS)表現出相對強度更高的CO脫附峰。脫附峰從325 ℃延伸至480 ℃,最大值在420 ℃左右,這可歸于較強的CO非解離吸附。研究中采用高斯函數擬合計算出脫附CO量,如表1所示。CO脫附量數據可以看出合成方法明顯影響了CO在催化劑表面的吸附能力。采用低溫等離子體法可以提高KNiMo基催化劑的CO非解離吸附量,其中,KNiMo-DPS催化劑具有最高的CO吸附量,為75 μmol/g。
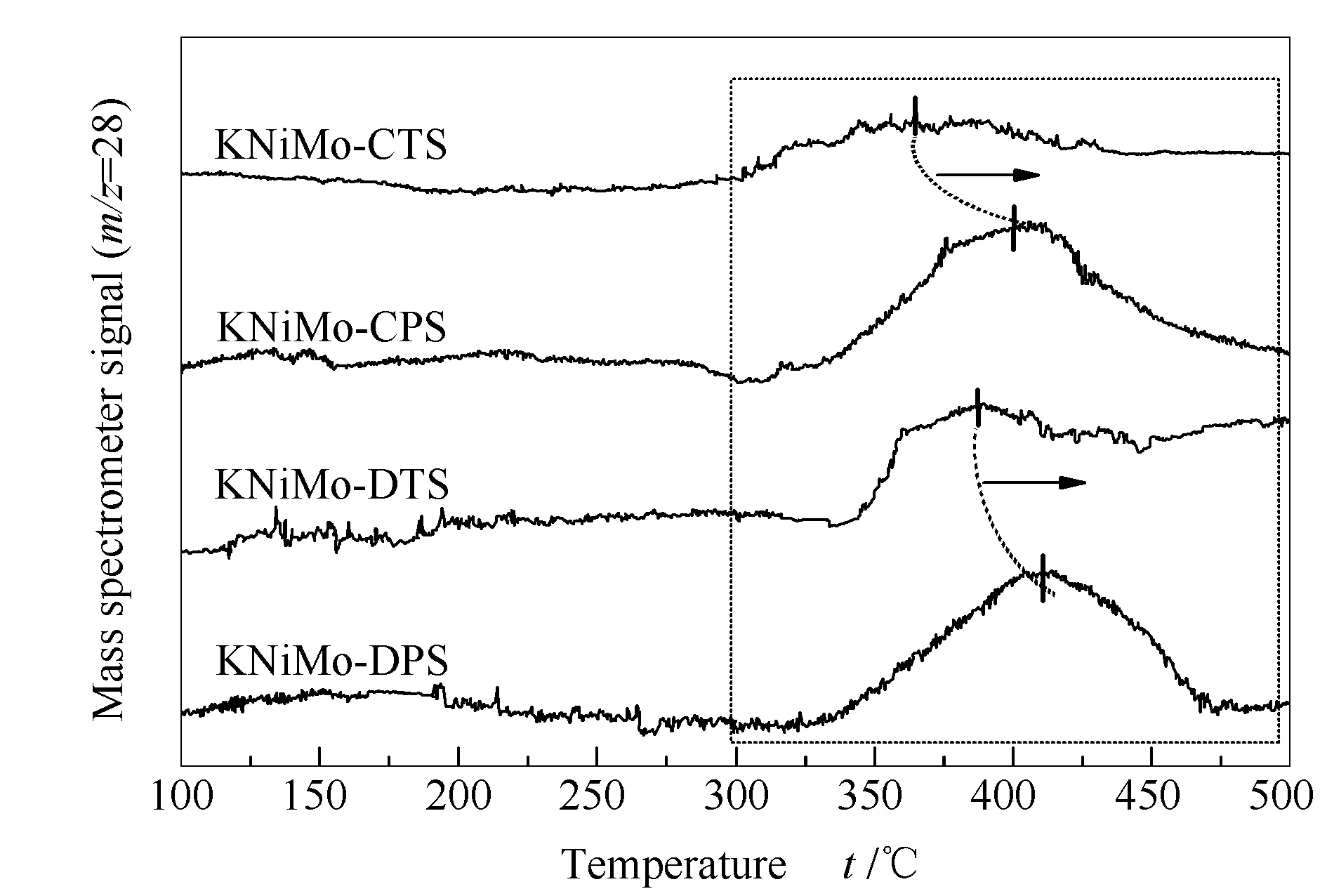
圖 6 不同方法所合成KNiMo基催化劑的CO-TPD譜圖
2.5 原位CO吸附DRIFTS分析
圖7為KNiMo-DPS催化劑的原位CO吸附DRIFTS譜圖。由圖7可知,所有紅外吸收譜圖均在2050-2250和2300-2400 cm-1處出現特征紅外吸收峰,分別歸屬于CO和CO2的振動吸收[40,41]。從圖7可以發現,催化劑上所吸附CO的振動吸收峰在2180和2118 cm-1左右[40],與文獻中氣態CO相比(彎曲振動吸收峰2200 cm-1,伸縮振動吸收峰2140 cm-1[42]),催化劑表面吸附CO振動吸收峰具有相對較低的波數,這也說明催化劑表面有利于CO非解離吸附,與CO-TPD分析結果一致。同時也觀察到CO2振動吸收峰產生于250 ℃左右。如文獻中所指出,Mo基催化劑有利于CO的解離和烴類物質的生成,所形成的C*物種與吸附的氫反應,轉化為CH2物種。這也表明,C*和O*物種是可以在Mo基催化劑作用下通過CO解離而形成[43]。根據原位CO吸附DRIFTS分析,上述C*和O*物種在催化劑表面可以進一步與非解離CO作用從而得到CO2。通常因Mo基催化劑常用于催化水煤氣變換反應,所以水煤氣變換反應也被認為是HAS中CO2形成的主要途徑[44]。而CO原位紅外吸附譜圖分析結果證明,CO解離和隨后C*與CO的作用是形成CO2的另一條反應途徑。此外,實驗中隨著反應溫度的升高,CO2峰強度的增強也顯示出CO解離反應取決于反應溫度。
2.6 不同KNiMo基催化劑的低碳醇合成反應性能
對不同合成方法制備的KNiMo基催化劑進行了HAS反應活性評價,結果見表2。KNiMo-DPS、KNiMo-CPS、KNiMo-DTS和KNiMo-CTS催化劑的CO轉化率分別為32.3%、28.4%、21.7%和19.8%。對比不同方法所合成催化劑的CO加氫活性可以發現,采用低溫等離子體法一步合成的KNiMo-DPS催化劑其CO轉化率是傳統熱法合成的KNiMo-CTS催化劑的約1.7倍,是直接熱法合成的KNiMo-DTS催化劑的約1.5倍。由此可以看出,與傳統熱法合成的催化劑相比,低溫離子體法不僅制備時間短、條件溫和,而且所制備的KNiMo催化劑具有更高的CO加氫反應活性。前述表征分析結果表明,低溫等離子體法制備的催化劑,如表1所示KNiMo-DPS的比表面積比KNiMo-CPS大。由于后者比前者含有一步熱焙燒處理過程,而高溫熱處理會導致顆粒的團聚,因此,降低了比表面積,從而使活性位數量和活性相的分散度下降,所以相較KNiMo-DPS催化劑,KNiMo-CPS催化劑的催化活性較低。此外,由于化學和物理性質的微小差異,熱合成的KNiMo-DTS和KNiMo-CTS催化劑表現出類似的催化活性。與傳統熱法合成的催化劑相比,低溫等離子體法合成的KNiMo基催化劑表現出更高的CO加氫反應活性。根據H2-TPD表征分析(請參見圖5、表1),使用不同的合成方法所制備的KNiMo基催化劑上吸附氫量明顯不同。KNiMo-DPS催化劑具有最大的氫吸附量和最高的CO轉化率。這些結果意味著活性氫吸附量與CO加氫反應活性呈現正相關性:吸附氫物種越多,對應催化劑的CO加氫活性越高。同時,低溫等離子體合成的KNiMo基催化劑具有高CO轉化率亦歸因于活性相無序且高度分散,這種結構提供了大量的配位不飽和活性位點以吸附和活化反應物分子。根據TEM照片可知,等離子體合成的催化劑表現出粒徑小(小于10 nm)、堆疊層數低(1-3層)的結構特征(參見圖3)。這些薄而短的片層可以增加邊、角位與基面位的比例,從而暴露出更多的活性位點,增加催化活性中心數量。
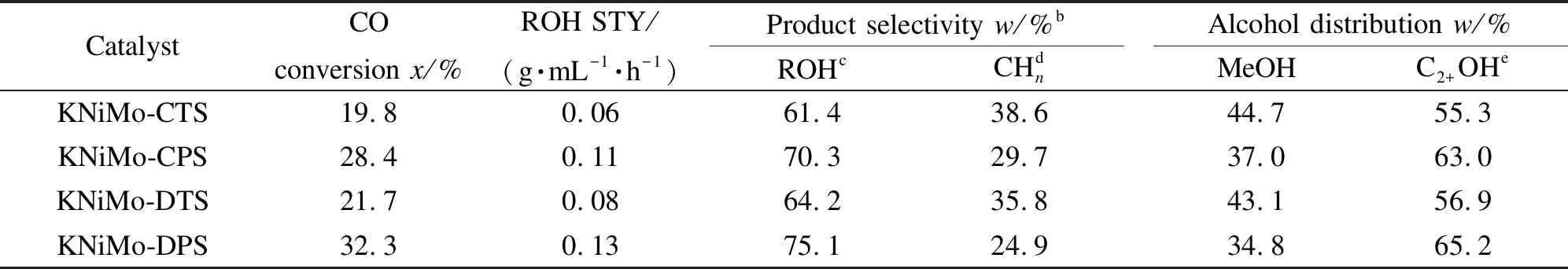
表 2 不同合成方法制備KNiMo基催化劑的HAS催化反應性能a
note: reactions were carried out at 350 ℃, 5.0 MPa, GHSV = 5000 h-1, H2/CO = 2. ROH means total alcohols and CHnis total hydrocarbons. STY is space-time yield and the product selectivity is CO2free-based. Alcohols with carbon number of 2-5 (mainly ethanol, 1-propanol, 1-butanol, 1-pentanol and 2-propanol) were obtained in the product
由表2可知,不同催化劑的總醇選擇性順序依次為KNiMo-DPS(75.1%) > KNiMo-CPS(70.3%) > KNiMo-DTS(64.2%) > KNiMo-CTS(61.4%)。前期已報道的HAS研究表明,非解離活化的CO對于CO加氫成醇過程是必不可少的[45,46]。根據圖6和表1所示CO-TPD表征結果,KNiMo-DTS和KNiMo-CTS催化劑對CO非解離活化的能力相對較低,因此,會導致總醇選擇性的下降。而低溫等離子體法合成的KNiMo基催化劑(KNiMo-DPS和KNiMo-CPS)具有高的非解離CO吸附能力,這表明低溫等離子體法制備的KNiMo基催化劑更有利于CO的非離解活化和插入反應,因此,其總醇選擇性相對更高。
與此同時,低溫等離子體合成的KNiMo基催化劑顯示出高的C2+醇選擇性。其中,KNiMo-DPS催化劑對C2+醇的選擇性最高,達到65.2%,ROH的STY(時空產率)為0.13 g/(mL·h)。這一實驗結果可以通過以下幾方面來解釋:在CO加氫制低碳醇研究中,CO插入機理被廣泛接受[39],Mo基催化劑上的HAS既需要在催化表面上的解離吸附CO,又需要非解離吸附CO。NiMoS相可以催化碳鏈生長和CO解離,而K助劑分散在其表面用以促進CO插入反應。各物種之間的協同作用對于低碳醇的合成是必不可少的。圖4所示的HAADF-STEM照片表明,K、Ni、Mo元素均勻分散并且相伴共存,這說明采用低溫等離子體法可以制備出具有良好結構穩定性的均勻分散KNiMo基催化劑。各元素的均勻分散共存與緊密接觸有利于其之間的協同催化作用,從而提高了C2+醇選擇性。
考察了低溫等離子體法KNiMo基催化劑的反應穩定性,具體見圖8。
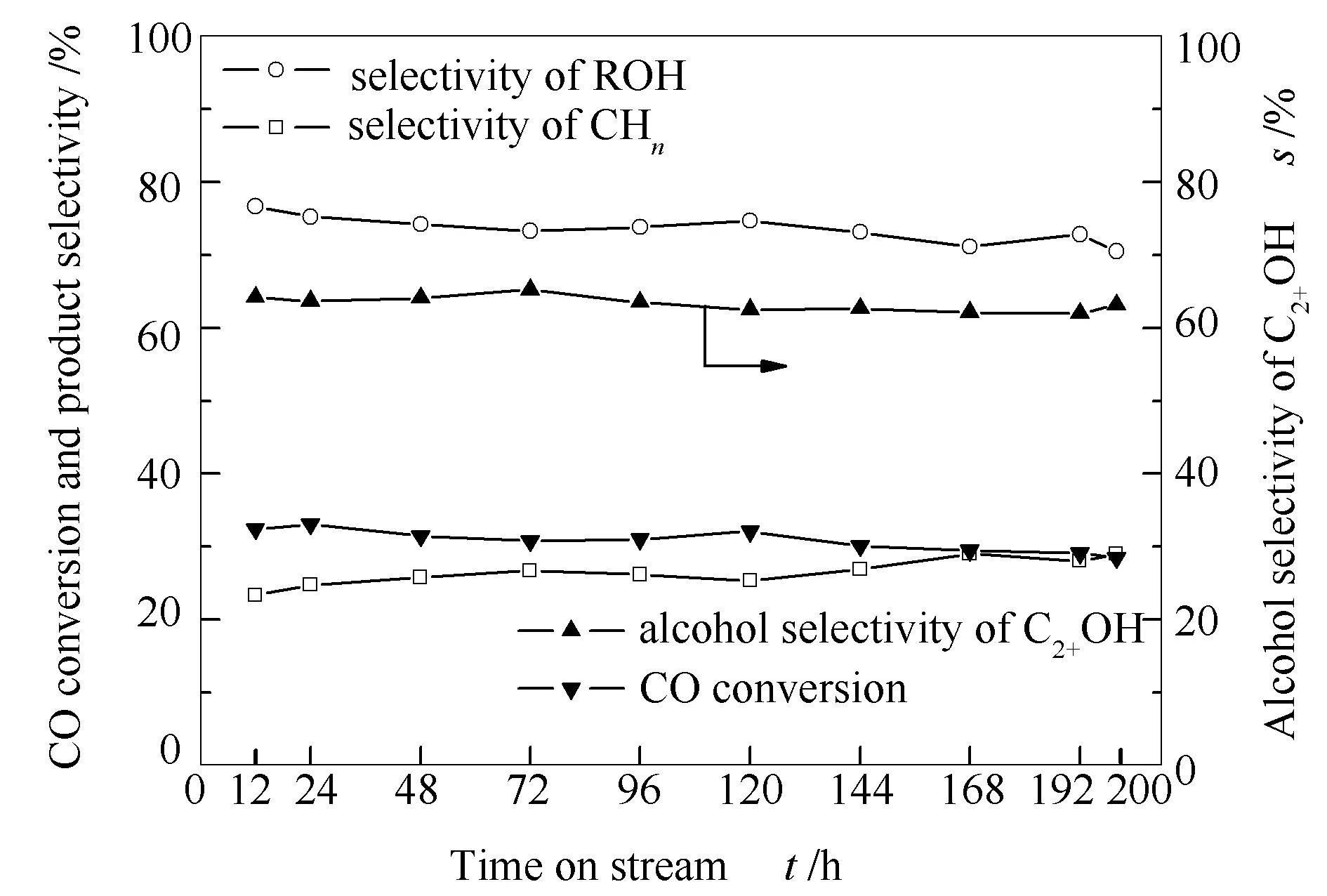
圖 8 KNiMo-DPS催化劑的穩定性
由圖8可知,200 h在線反應低溫等離子體合成的KNiMo基催化劑的催化性能穩定,未出現催化劑失活的現象。可以確定低溫等離子體法制備的催化劑具有良好的穩定性,也進一步說明各物種之間的協同作用在CO加氫反應過程中保持良好。
圖9(a)為KNiMo-DPS催化劑長周期反應前后的XRD譜圖。由圖9(a)可知,經長時間反應后的催化劑與反應前對比無晶相變化。根據Scherrer方程計算發現長周期反應后顆粒平均粒徑由7.1 nm增大至9.3 nm左右。這也證明活性相在反應過程中具有優異的穩定性。以上結果表明,KNiMo催化劑在CO加氫制低碳醇反應體系中可以穩定存在,并保持良好的反應性能。此外,如圖9(b)所示,傳統熱法制備的KNiMo催化劑在反應達到穩態后也未存在新物相的生成,這一結果與文獻報道的結果相一致[47],即Mo基催化劑在CO加氫反應中可以穩定存在。
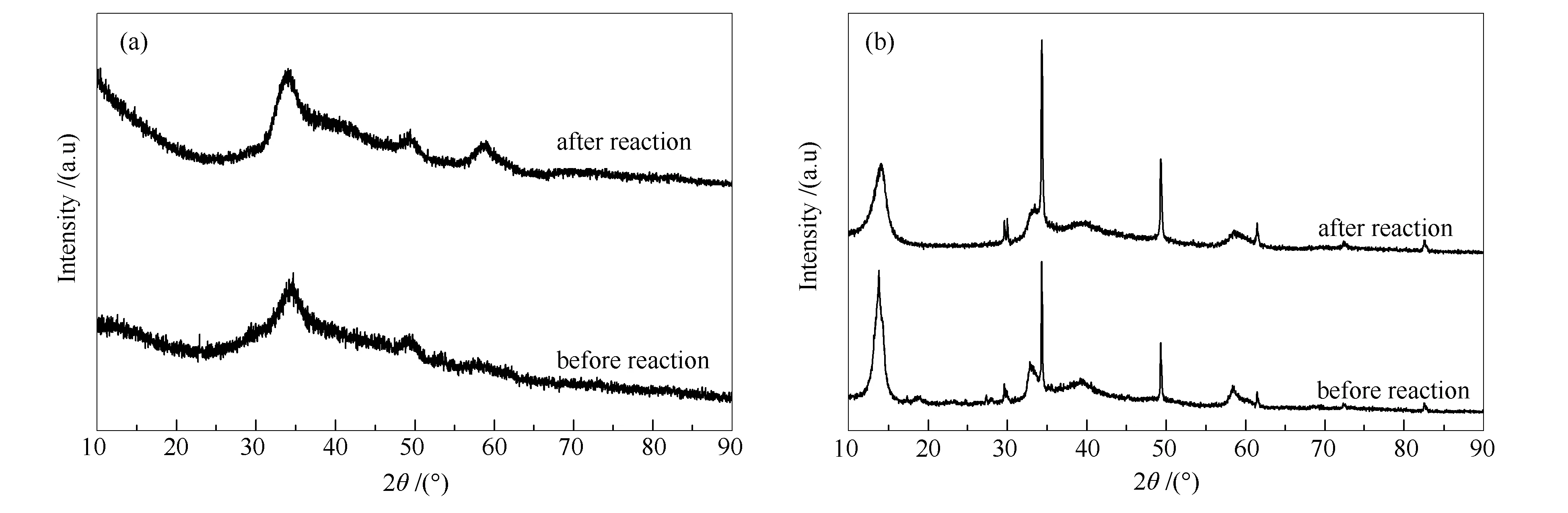
圖 9 KNiMo-DPS催化劑(a)和KNiMo-CTS催化劑(b)反應前后的XRD譜圖
3 結 論
采用低溫等離子體法在溫和條件下合成了高性能KNiMo基催化劑。催化劑具有更薄、更短的片層結構并暴露出更多邊、角位的催化活性位,促進了CO加氫活性和低碳醇的生成。與傳統熱法相比,低溫等離子體法不僅縮短了制備時間,還合成出層數少、粒徑小的高分散KNiMo基催化劑。其中,KNiMo-DPS催化劑表現出最優的HAS催化反應性能。在5 MPa、350 ℃、空速5000 h-1的反應條件下,CO轉化率達到32.3%,總醇選擇性75.1%,其中,總醇中C2+醇的選擇性為65.2%。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
