控制性打開二硫鍵-葡聚糖修飾對大豆分離蛋白表面活性性能及結構的影響
2019-06-19 02:20:48王碧璇李軍生鐘新閻柳娟黃國霞
中國飼料 2019年11期
關鍵詞:大豆
王碧璇 , 李軍生 *, 鐘新 , 閻柳娟 , 黃國霞
(1.廣西科技大學生物與化學工程學院,廣西柳州 545006;2.廣西糖資源綠色加工重點實驗室,廣西柳州 545006;3.廣西高校糖資源加工重點實驗室,廣西柳州 545006)
大豆分離蛋白作為重要的植物蛋白來源,具有豐富的營養價值以及廣泛的功能特性,其中,乳化性以及起泡性是大豆分離蛋白最重要的功能之一。隨著食品工業的發展,提高表面活性變得至關重要,目前通過物理、化學、酶等改性方法,改善大豆分離蛋白的表面活性。王松等(2014)探討了大豆分離蛋白與葡萄糖的最優條件,制備了大豆分離蛋白-葡萄糖的復合產物,最終得到產物的乳化活性是大豆分離蛋白的3.94倍,Wang等(2013)添加了乳糖進行糖基化修飾,發現其乳化性和乳化穩定性是未經過修飾的2.32倍和2.67倍,由此說明糖基化可以有效改善大豆分離蛋白的表面活性。
受蛋白質特殊結構的限制,大部分的疏水性氨基酸被包裹在分子內部,因此大多數糖基化發生在分子的表面。本研究采用綠色的氧化劑,對蛋白質進行氧化,將蛋白質內部二硫鍵的非共價力作用控制性的斷開,使得內部的疏水性基團暴露在分子的表面。在此基礎上,引入葡聚糖分子。通過對大豆分離蛋白的結構、性能進行分析,可以為大豆分離蛋白的開發和利用提供更好的思路,并且為大豆蛋白改性提供新的方法。
1 材料與方法
1.1 材料與試劑 豆粕,市售;葡聚糖購自上海源葉生物有限公司;鄰苯二甲醛購自國藥試劑;其他試劑均為分析純,購于西隴化工股份有限公司。
1.2 儀器與設備 高速冷凍離心機(美國貝克曼庫爾特有限公司);紫外可見分光光度計(Agilent Technologies);熒光分光光度計(日本島津公司);馬爾文納米粒度和Zeta電位分儀(英國馬爾文儀器有限公司);電泳儀(美國Bio-Rad伯樂公司)。
1.3 試驗方法
1.3.1 大豆分離蛋白的制備 將購得的豆粕研磨,過80目篩。得到的蛋白粉按照重量體積比1:10溶于去離子水中。用2 mol/L的NaOH調節pH至7.0,攪拌1 h,懸浮液以8000 r/min離心30 min。得到的上清液用2 mol/L的HCl調節pH至4.5,5000 r/min離心30 min,得到的蛋白質溶液用冷凍干燥機干燥,研磨成大豆分離蛋白粉,待用。以上所有的工作均在室溫下完成。
1.3.2 大豆分離蛋白的氧化 將1.25 g大豆分離蛋白溶于50 mL緩沖溶液當中,依次加入0、0.1、0.2、0.5、0.7、0.9 mL 過氧乙酸,得到的混合物置于4℃的黑暗環境中反應,過夜。
1.3.3 大豆分離蛋白糖基化反應 取一定量的大豆分離蛋白進行氧化,按蛋白質與糖1∶2的量進行反應,蛋白質、葡聚糖混合后磁力攪拌10 min,加入到50 mL具塞試管中,在100℃下加熱,反應1 h,反應結束后,立即放入冰水浴中冷卻,待測(或者冷凍干燥)。
1.3.4 二硫鍵含量的測定 采用 Ellman(1959)法稍作修改;配制濃度為0.1%DTNB,以及 0.1%NTSB試劑。測游離巰基時,取0.4 mL待測樣品,加入0.4 mL DTNB,用pH 8.0的磷酸鹽緩沖溶液定容至10 mL。當測定總巰基時,將DTNB替換為NTSB即可。在室溫下反應20 min,利用紫外分光光度計在412 nm處,測定吸光度。同樣的方法,配制不同濃度的谷胱甘肽,作標準曲線,得到的標準曲線為:
y=0.3697x-0.0162 R2=0.9976
式中:x為蛋白質質量濃度,mg/mL;y為412 nm處吸光度值。
還原型谷胱甘肽的摩爾質量為307.32 g/mol,因此巰基濃度(mol/L)的計算公式為:

1.3.5 大豆分離蛋白-葡聚糖復合產物接枝度的測定 采用OPA法測定自由氨基 (Cmj等,2003):OPA 的配制(此試劑現配現用),取 40 mg鄰苯二甲醛溶解于1 mL甲醇溶液中,加入20%(m/m)的 SDS(十二烷基硫酸鈉)2.5 mL,再加入0.1 mol/L 的硼砂 25.0 mL,加入 100 μL β-巰基乙醇定容至50 mL。
自由氨基的測定:取4.0 mL OPA于試管中,加入200 μL樣品,混合均勻,放在35℃的水浴鍋中加熱,取4.0 mL OPA,加入200 μL水作為空白,在340 nm處測其吸光度。同樣的方法,用賴氨酸作標準曲線。由標準曲線計算游離氨基的含量。

1.3.6 復合產物褐變度的測定 取0.1 g樣品,溶解于緩沖溶液當中,取1 mL加入5 mL的1%SDS,以緩沖溶液為空白,測定在420 nm處的吸光度值,以吸光度值大小表示樣品的褐變程度(Fogliano等,1999)。
1.3.7 起泡性和起泡穩定性的測定 取10 mL待測溶液定容至50 mL,用10000 r/min的速度在高剪切混合頭下攪打1 min,將均質后的溶液倒入100 mL量筒中,記下0 min時刻的泡沫體積V1,30 min后記下泡沫體積V2,采用以下公式計算:
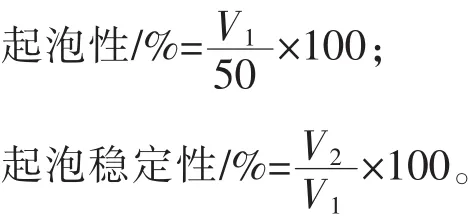
1.3.8 乳化性和乳化穩定性的測定 取15 mL待測溶液,加15 mL大豆油于燒杯中,以10000 r/min的速度在高剪切混合頭下攪打1 min,在0 min,30 min中在燒杯底部取20 μL樣品,與5 mL 0.1%的SDS混合,利用紫外分光光度計在500 nm處,測定其吸光度值(Molina等,2001):
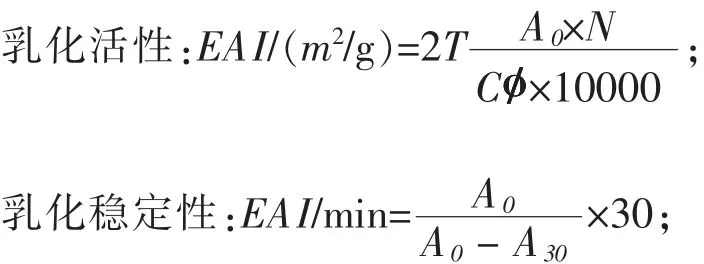
式中:T=2.303;N為蛋白質稀釋倍數;C為待測樣品蛋白質濃度,g/mL;o/為乳化液中油相所占體積分數,0.5;A0為 0 min的吸光度值;A30為30 min的吸光度值。
1.3.9 熒光光譜的測定 以290 nm為激發波長,將樣品以4000 r/min離心10 min,取0.1 mL上清液定容至10 mL,測定大豆分離蛋白各個樣品的內源熒光,雙縮脲法測定樣品的蛋白質含量。設定激發狹縫寬度:5 nm;發射狹縫狹縫寬度:3 nm;掃描范圍:300~ 420 nm;強度范圍:0~1000;掃速:中速。
1.3.10 SDS-PAGE的測定 參考Laemmli等(1970)的方法稍作修改,其中分離膠濃度13%,濃縮膠濃度5%。電泳條件:先采用90 V電泳30 min,后采用120 V電泳處理2.5 h。考馬斯亮藍R-250染色,而后脫色拍照。
2 結果與分析
2.1 二硫鍵打開程度與接枝度結果分析 糖基化反應是通過美拉德反應,其中蛋白質分子游離的氨基與多糖分子的羰基反應,連接到多糖分子的末端。一般采用接枝度,來評價糖基化的反應程度(張晉博,2013)。大豆分離蛋白與葡聚糖的糖基化產物的接枝度,如圖1所示,當二硫鍵打開率為46%時,接枝度明顯增加,由此可以斷定通過氧化控制性的斷開二硫鍵,可以有效提高糖基化反應的效率。
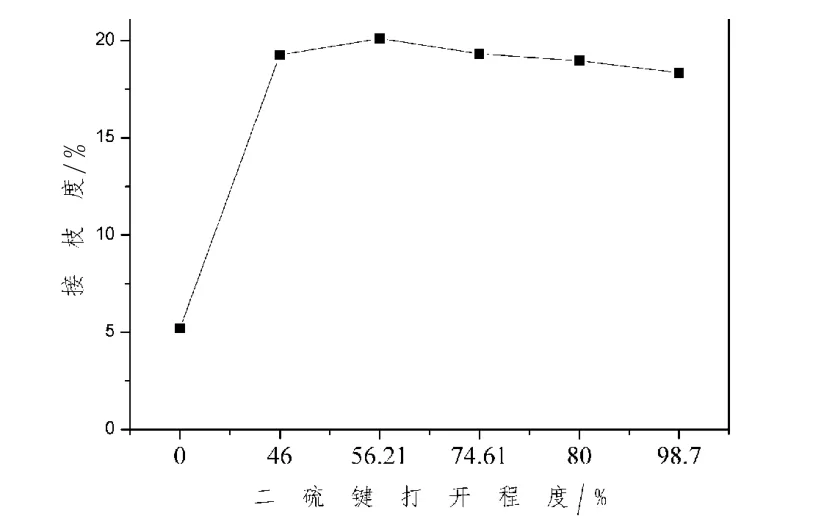
圖1 二硫鍵的打開程度對接枝度的影響
2.2 褐變度結果分析 糖基化反應的過程中,通常會伴隨著褐變現象的發生,產生的物質稱為類黑素。其中類黑素是由蛋白質中的氨基酸與糖分子的主干組成的。隨著反應的進行,產物的顏色會加深,因此可以通過反應產物的顏色,間接的反映接枝度的大小。因此測定接枝產物的褐變程度,更能直觀的反映美拉德反應的程度。如圖2所示,褐變程度與接枝度的趨勢相符,進一步證明了復合修飾可以提高糖基化效率。
2.3 起泡性以及起泡穩定性結果分析 圖3為二硫鍵的打開程度對起泡性以及起泡穩定性的影響,起泡性依次為 110%、180%、200%、195%、180%、170%,起泡穩定性為 30.77%、45.83%、50.77%、55.56%、57.78%、56.29%。隨著二硫鍵打開程度的增加,起泡性出現先增加后降低的趨勢,這是由于二硫鍵的斷開,蛋白質內部的疏水性基團暴露在分子的表面,蛋白質在溶液狀態時親水基團與疏水基團比例接近平衡,蛋白質分子在溶液表面的定向排列更加有序,因此起泡性增加。而隨著二硫鍵斷開程度的增加,結構遭到破壞,因此出現降低的趨勢。
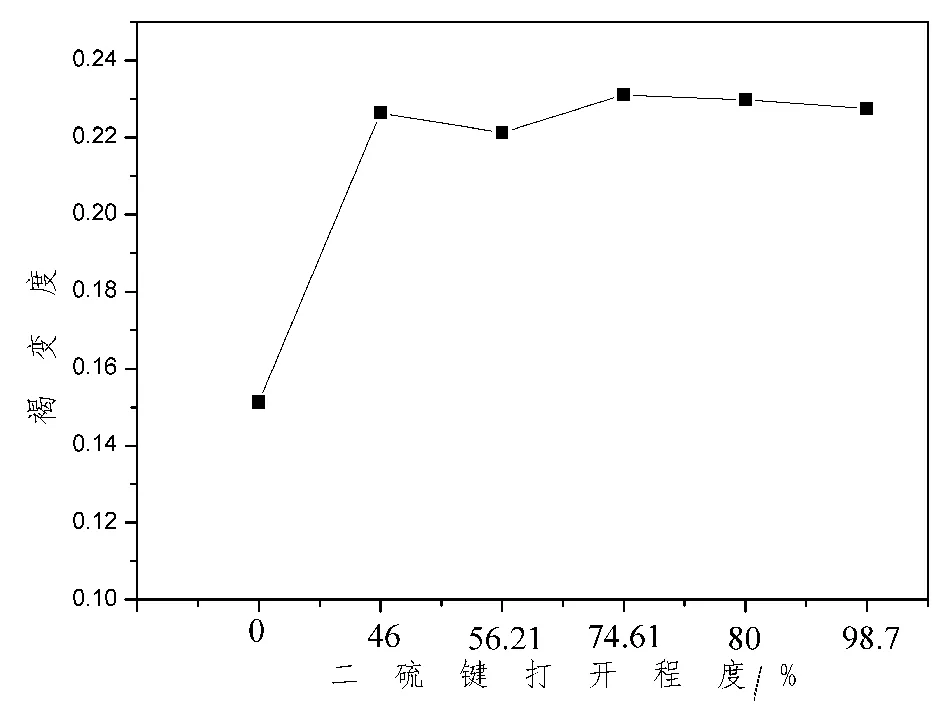
圖2 二硫鍵打開程度對褐變度的影響

圖3 二硫鍵打開程度對起泡性以及起泡穩定性的影響
如圖4所示,起泡性依次為154%、158%、202%、186%、190%、130%,起泡穩定性依次為66.37%、69.34%、71.05%、82.35%、80%、79.5%,與圖3比較,起泡性與起泡穩定性有較大的提高。蛋白質與葡聚糖發生糖基化反應,有較高的溶解性,可以比蛋白質以更快的速度轉移到分子的界面,以此來提高大豆分離蛋白的表面活性。蛋白質的起泡穩定性主要是由于蛋白質在界面處形成吸附層,穩定了泡沫并且可以防止泡沫的破裂。泡沫的體積隨著放置時間的增加而減少,而通過復合修飾后可以明顯改善起泡的穩定性。糖基化反應可以改善大豆分離蛋白的發泡能力,降低表面張力,改變樣品的溶解度、表面疏水性、泡沫穩定性等。
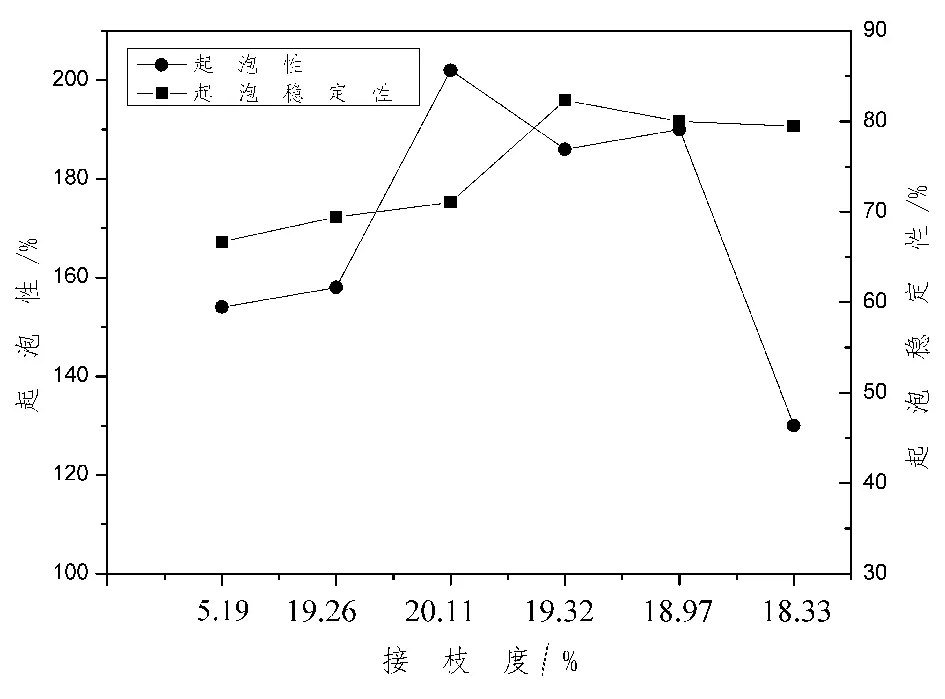
圖4 接枝度對起泡性以及起泡穩定性的影響
2.4 乳化性以及乳化穩定性結果分析 不同程度的打開二硫鍵,乳化性依次為15.93、25.19、24.73、22.05、23.7、21.44 m2/g,乳化穩定性依次為99.11、140.48、138.44、127.79、132.41、111.33 min(圖5);而復合修飾樣品的乳化性依次為33.07、33.52、31.66、26.76、25.01、24.54 m2/g, 乳化穩定性 依 次 為 169、273、474、457、452、236 min (圖6),從數值上來看,復合修飾的樣品具有更好的乳化性和乳化穩定性。
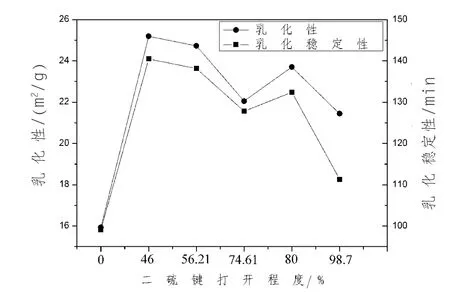
圖5 二硫鍵打開程度對乳化性以及乳化穩定性的影響
從圖6中可以看出,混合物的乳化性和乳化穩定性得到改善,均出現先升高后降低的趨勢,當接枝度為19.26%時,乳化性能達到最高為33.52 m2/g,此時的穩定性并非最好,穩定性最高時,乳化性開始下降,這是由蛋白質緊密的三級結構引起的,由熒光光譜分析得到了證實。作為非表面活性劑的親水性多糖,葡聚糖在空氣-水界面上不具備吸附能力,但作為增稠劑或凝膠劑能顯著提高蛋白質泡沫的穩定性。如預期的一樣,大豆分離蛋白與葡聚糖聚合物的乳化性和發泡性得到改善,打開二硫鍵使得美拉德反應進一步增強。大多數的疏水性基團和賴氨酸殘基被包裹在分子的內部,未折疊蛋白質的賴氨酰殘基暴露于外部,相比較折疊的蛋白質有較小的空間位阻,因此容易與多糖中的還原羰基發生反應。Nakamura(1998)等指出,乳化性增加的原因可能是多糖鏈附著于蛋白質/肽抑制了每個油滴或蛋白質/肽之間的聚集。另一方面,用于糖基化的多糖研究是支鏈和長鏈糖類,這提供了更多的空間位阻,預防脂肪小滴的聚結。
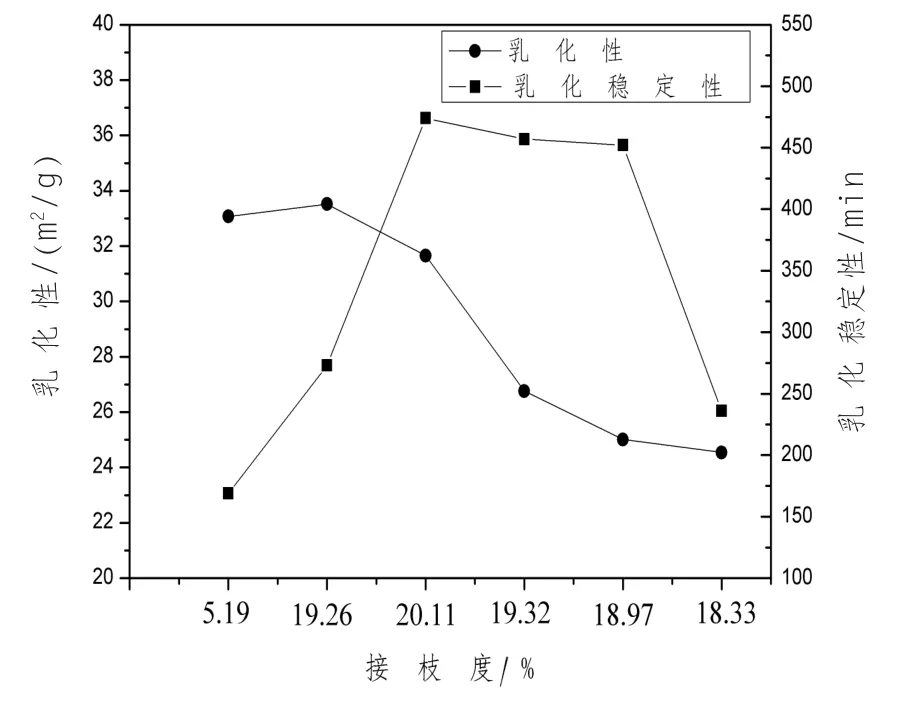
圖6 接枝度對乳化性以及乳化穩定性的影響
2.5 熒光光譜結果分析 從圖7可以看出,天然的大豆分離蛋白在330 nm處,有最大吸收峰。當二硫鍵斷開程度為46%時,峰高最強,發生位移的程度也最大,說明所具有的疏水性程度最高,隨著過氧乙酸量的增加,最大吸收峰逐漸變小,可能是由于從蛋白質分子發生聚集,使得發色基團從蛋白質分子的外部遷移到蛋白質分子的內部。圖8表明,加入葡聚糖分子后,樣品的吸收峰下降明顯,并且氧化后樣品的吸收峰下降的幅度大于未氧化糖基化樣品的吸收峰,并且最大吸收峰發生了藍移。
由于過氧乙酸的氧化,使得蛋白質分子的結構變得疏松,內部的極性基團暴露出來,因此葡聚糖的加入,更容易發生美拉德反應。隨著過氧乙酸濃度的增加,暴露出更多的極性基團,糖基化速率增加,導致了熒光發色基團的猝滅,甚至消失。
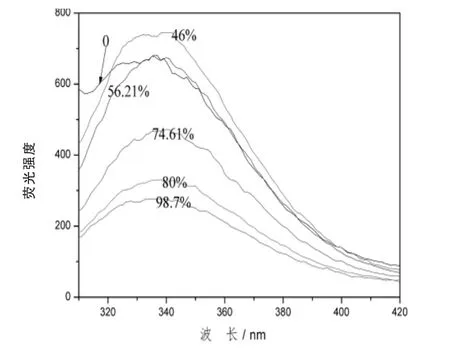
圖7 二硫鍵打開程度對內源熒光強度的影響
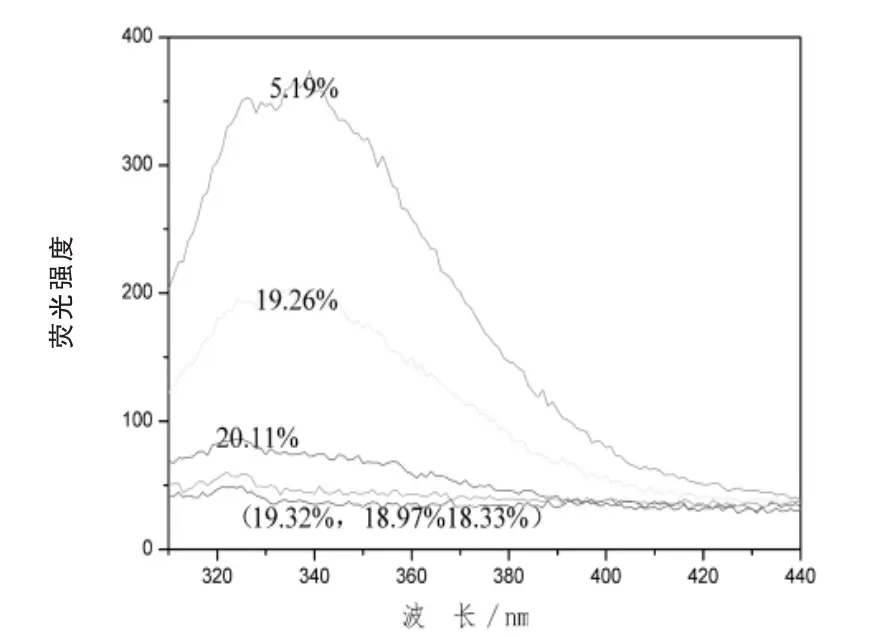
圖8 接枝度對內源熒光強度的影響
2.6 SDS-PAGE結果分析 從圖9可以看出,對于未經過過氧乙酸氧化的大豆分離蛋白,AS條帶組有三條條帶,中間條帶顏色較深且較寬。二硫鍵依次被斷開的過程中,AS條帶由窄變寬,并且顏色逐漸變深,位置與未經氧化的1比較,上移;說明AS條帶存在二硫鍵,被氧化斷開;Band1隨氧化過程的增加逐漸出現,Band2隨氧化程度增加而逐漸變淺甚至消失。這一現象說明,過氧乙酸的氧化作用強于還原作用,可以將蛋白質分子中的二硫鍵不同程度上斷裂,其次,經氧化后的蛋白質樣品條帶顏色由淺變深,這意味著在二硫鍵打開程度較小時亞基發生解離和條帶分散,結構變得疏松(盧巖等,2014)。
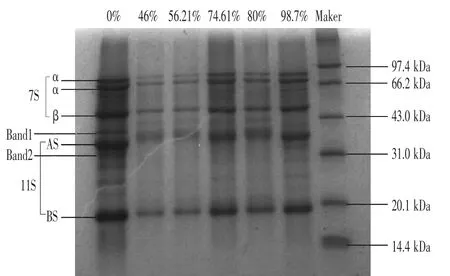
圖9 二硫鍵打開程度不同SDS凝膠電泳圖
從圖10中可以看出,為斷開二硫鍵,加入葡聚糖時,對天然大豆分離蛋白樣品的條帶、種類沒有明顯的影響,說明葡聚糖對大豆分離蛋白的結構影響甚微。而在氧化的基礎上,加入葡聚糖,條帶變化比較明顯。隨著氧化的增加,接枝度增加,A組條帶上移,并且條帶變寬,說明大豆分離蛋白與葡聚糖反應,生成了新的產物,導致分子量增加;α'和α亞基,在未加入過氧乙酸時,顏色就明顯變淺,說明7S中不含二硫鍵,這與Adachi等(2003)的研究結論相符。因此在未氧化時,7S亞基最先消失,斷開二硫鍵后,11S的酸性亞基,逐漸消失,由此也可以說明過氧乙酸斷開二硫鍵后,可以加速11S酸性亞基與葡聚糖發生糖基化反應。這與Mu等(2011)的報道一致,糖基化反應過程中7S亞基最先消失,其次是11S的酸性亞基,可能是因為11S的堿性亞基含有少量的賴氨酸和精氨酸等。
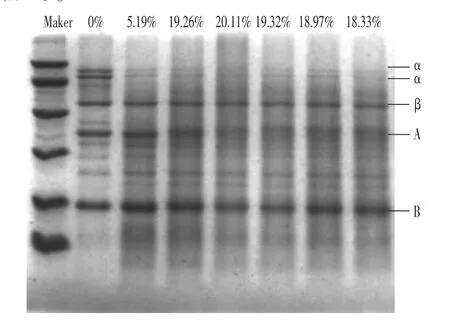
圖10 接枝度不同SDS凝膠電泳圖
通過對SDS-PAGE圖的分析,一方面證明有大豆分離蛋白與葡聚糖發生了美拉德反應,有糖基化反應的生成;另一方面說明過氧乙酸氧化斷開二硫鍵可以有效提高糖基化反應的速率。
3 結論
大豆蛋白從理論上講,有良好的表面活性,并且其來源廣泛,價格便宜,因此制備大豆蛋白基表面活性劑變得可行。本試驗采用過氧乙酸打開蛋白質分子中的二硫鍵,使得蛋白質發生水解反應,在肽鏈解旋,重新排列的基礎上,引入葡聚糖分子,使其發生糖基化反應,進一步提升大豆分離蛋白的表面活性劑。對其復合產物的乳化性、起泡性以及相對應的穩定性進行了測定,發現表面活性有很大的提升,為制備大豆分離蛋白基表面活性劑奠定了基礎。
猜你喜歡
農業科技通訊(2023年1期)2023-02-12 07:09:18
今日農業(2022年16期)2022-11-09 23:18:44
中國化肥信息(2022年7期)2022-08-31 01:29:28
中國化肥信息(2022年5期)2022-08-30 01:58:26
今日農業(2021年20期)2021-11-26 01:23:56
今日農業(2021年14期)2021-10-14 08:35:34
下一代英才(酷炫少年)(2018年6期)2018-07-09 03:17:44
農產品市場周刊(2017年4期)2017-03-03 19:40:05
兒童故事畫報·智力大王(2015年10期)2016-01-27 01:01:35
讀寫算(中)(2015年10期)2015-11-07 07:24:12
