測定飼料中維生素A和維生素E含量方法的研究
2019-06-19 02:20:52裘丞軍任玉琴
中國飼料 2019年11期
關鍵詞:方法
裘丞軍, 任玉琴
(浙江省獸藥飼料監察所,浙江杭州 311199)
維生素A和維生素E是維持動物體正常代謝所必需的脂溶性維生素,具有促進生長,提高免疫力等作用,已經廣泛應用于大規模畜禽養殖中。在飼料添加劑中主要以維生素A乙酸酯和dl-α-生育酚乙酸酯的形式存在,并且通過各種壁材包被保護(楊優敏,2016)。目前測定飼料中維生素A和維生素E的方法已有國家標準GB/T 17812-2008和GB/T 17817-2010,主要是皂化萃取法和甲醇直接提取法,還有堿性蛋白酶水解法(趙曉華等,2010)。皂化萃取法操作步驟繁瑣,時間長,消耗試劑多;甲醇直接提取法和酶解法結果不穩定,有些包被產品未能被有效提取。因此本研究擬建立一種能穩定、快速、準確、同時測定飼料中維生素A和維生素E的方法,通過氫氧化鉀溶液皂化,乙醇提取,將包被物質溶解,維生素A乙酸酯和dl-α-生育酚乙酸酯轉化為維生素A醇和dl-α-生育酚,用外標法計算含量。
1 材料與方法
1.1 試劑和儀器 維生素A乙酸酯標準品(德國Dr.Ehrenstorfer公司);維生素E乙酸酯標準品(Fluka公司);無水乙醇、抗壞血酸、冰乙酸、氫氧化鉀均為分析純;甲醇為色譜純;水是超純水;配合飼料、濃縮料、維生素預混料、復合預混料為日常檢測時收集。
Agilent1260高效液相色譜儀,紫外可變波長檢測器(美國Agilent公司);KQ-500E超聲儀(昆山超聲儀器公司);ZP205電子天平(Mettler公司)。
1.2 色譜條件 色譜柱:Agilent XDB-C18柱,柱長150 mm,內徑 4.6 mm,粒徑 5 μm;流動相:甲醇+水=98+2;流速:1.2 mL/min;進樣量:20 μL;柱溫:30 ℃;紫外檢測波長:0~7min,326nm;7~20min,280nm。
1.3 飼料樣品前處理 準確稱取飼料樣品適量(不大于 2 g,精確至0.1 mg),置于 50 mL容量瓶中,依次加入無水乙醇6 mL,10%抗壞血酸水溶液 2.0 mL,470 g/L氫氧化鉀水溶液 2.0 mL,混勻,置65℃水浴超聲波皂化反應20 min,取出,立即加入1.0 mL冰乙酸調pH至6.0~7.5,冷卻至室溫,用無水乙醇定容至刻度,混勻,靜置。
1.4 標準儲備溶液配制 精密稱取標準品維生素A乙酸酯62.19 mg,維生素E乙酸酯207.58 mg,分別置于100 mL棕色容量瓶中,加入無水乙醇超聲波助溶,冷卻至室溫,用無水乙醇定容至刻度,混勻,得到維生素A濃度為1807.8 IU/mL,維生素E濃度為2075.8 μg/mL,-20℃避光保存。
1.5 標準工作液配制 精密移取維生素A儲備液和維生素E儲備液適量,分別置于 50 mL容量瓶中,按照1.3方法處理,用無水乙醇稀釋定容成維 生 素 A 濃 度 0.03616、0.1808、0.3616、1.085、5.424、27.12、54.24、108.5 IU/mL 和維生素 E 濃度0.62274、1.2455、6.2274、12.455、24.910、62.274、124.55 μg/mL系列標準工作液。
2 結果與分析
2.1 色譜條件的確立 參考國家標準GB/T 17817-2010和GB/T 17812-2008,維生素A和維生素E的分析方法有正相和反相兩種色譜體系。正相法分離效果好,能夠有效分離異構體,并且試樣中溶解的脂肪容易被流動相沖洗出色譜柱,但是作為溶劑的正己烷揮發性比較大,長時間分析樣品濃度會變化,精密度相對較差;反相法穩定性高,對于飼料中維生素A和維生素E的分離效果好,保留時間適中(見圖1)。因此選擇反相色譜系統,采用 C18 柱,甲醇和水(98∶2)作為流動相,利用紫外檢測器可切換波長的功能在326 nm和280 nm處分別記錄維生素A和維生素E的色譜峰。
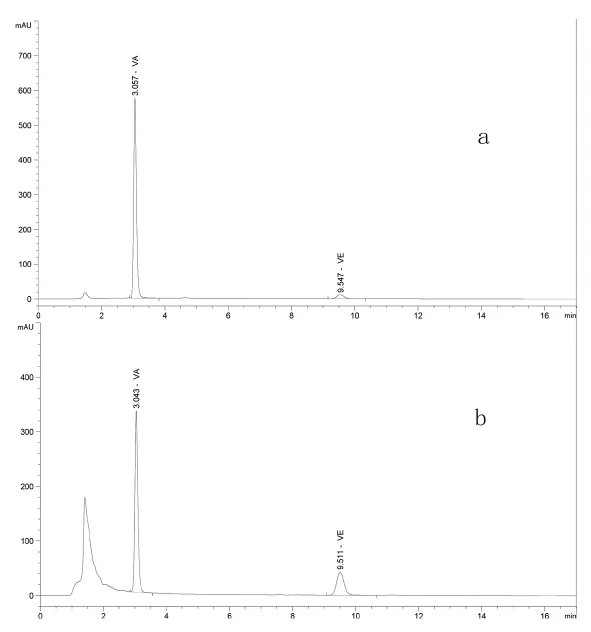
圖1 維生素A、維生素E標準圖譜(a)和樣品溶液圖譜(b)
2.2 提取劑的確定 本方法對比選用甲醇、乙醇作為提取劑,實驗表明乙醇比甲醇的提取效果好,同時對人體危害性小,因此選擇乙醇作為提取劑。
按照本方法,飼料樣品前處理過程分為皂化提取和乙醇稀釋定容兩部分。超聲波水浴加熱皂化反應時體系中水和乙醇的比例對于一部分飼料中維生素的檢出值影響比較大。尤其是水產料中的水分散性維生素顆粒,低含水量皂化液使得飼料黏稠不易分散,維生素A和維生素E檢出值相對較低;相反,濃縮料和配合料在水含量高時容易黏稠不易分散。實驗比較分析含水量分別為13%、20%、28%、40%、53%五種乙醇皂化體系,結果如表1,當皂化體系水分含量為40%和53%時,提取效果較好。
由于流動相是高比例甲醇體系,如果上機溶液水分含量較高,一些溶于水不溶于甲醇的物質在進樣時容易析出,從而污染色譜柱。用水分含量分別為5%、10%、16%三種上機乙醇溶液與甲醇1∶1混合,研究最終定容后上機溶液水分含量對于色譜系統的影響。發現16%溶液產生渾濁,10%和5%溶液澄清。
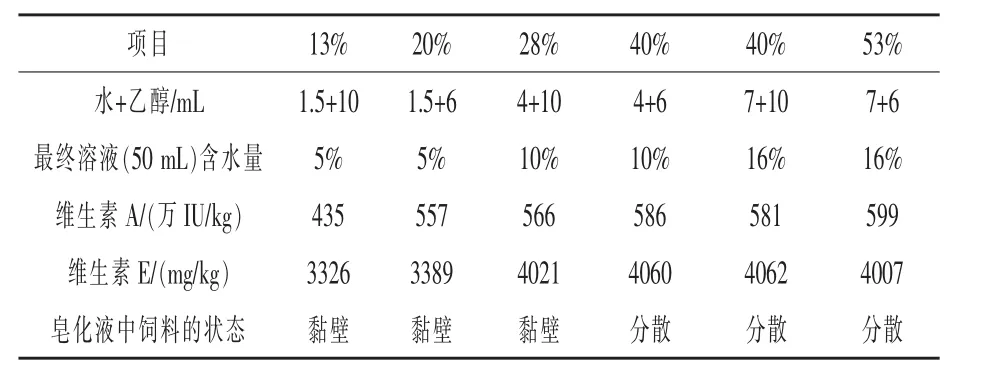
表1 某水產維生素預混料(水分散型)皂化體系比較
綜合分析,確定皂化時水含量為40%,最終定容上機液水分含量為10%,即在50 mL容量瓶中,加入無水乙醇6 mL,抗壞血酸水溶液2.0 mL,氫氧化鉀水溶液2.0 mL,然后皂化提取,加入1.0 mL冰乙酸,冷卻至室溫,用無水乙醇定容至刻度。
2.3 抗氧化劑選擇 在高溫強堿的環境中維生素E特別容易被氧化,因此在皂化提取時一般加入抗壞血酸(張艷海等,2015;謝云峰等,2014;黃誠等,2012)或者沒食子酸(李頌群等,2013)防止維生素氧化,國家標準GB/T 17812-2018和GB/T 17817-2010中還加入了2,6二叔丁基對甲酚(BHT)。本方法采用抗壞血酸作為抗氧化劑,在皂化提取液中分別加入2~200 mg的量,其他條件按照1.3方法處理,比較維生素A和維生素E的值。結果如表2,當抗壞血酸少于40 mg時,維生素E全部或部分損失,維生素A部分損失;當加入量大于100 mg時,維生素A和維生素E的值相對穩定,本方法選取抗壞血酸加入量為200 mg,即2.0 mL10%的抗壞血酸水溶液。
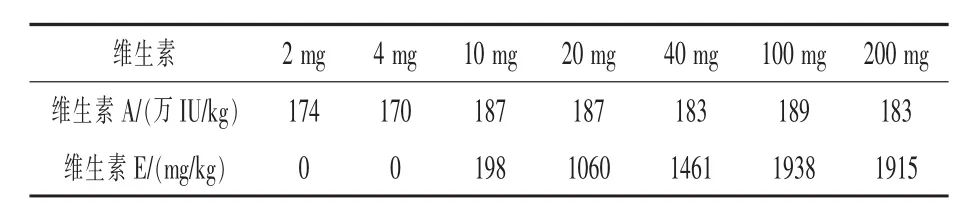
表2 抗壞血酸含量對維生素測定結果的影響
2.4 皂化提取加堿量控制 取5份相同飼料,分別加入1.5~60 mmol不等的氫氧化鉀,其他條件按照1.3方法,分析色譜峰。結果如表3,隨著堿含量的增加,皂化液pH不斷升高,當氫氧化鉀低于3.0 mmol時,維生素E未皂化或未完全皂化,維生素A未完全皂化;當氫氧化鉀大于10 mmol時,維生素A和維生素E完全皂化,并且值相對穩定。因此,為保證皂化效果,選擇加入2.0 mL 470 g/L氫氧化鉀(約16 mmol),最后皂化完成時用1.0 mL冰乙酸調pH為6.0~7.5。
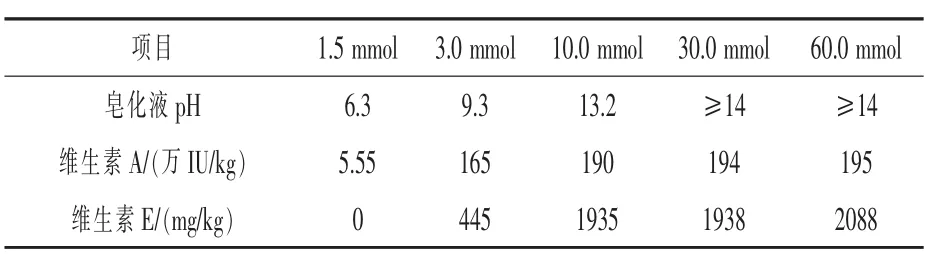
表3 不同堿含量對維生素測定結果的影響
2.5 皂化溫度控制 取5份相同的飼料,分別在50~100℃的水浴中超聲加熱,其他條件按照1.3方法,分析色譜峰,計算維生素A和維生素E的含量。結果如表4,維生素A在60~70℃時含量最大,低于60℃或高于70℃含量都偏低;維生素E在50~70℃時含量較穩定,80℃以上時含量下降。分析原因,維生素A在高溫下會發生異構和聚合反應(俞安,2012),維生素E在堿性液體高溫條件下易氧化降解;同時維生素包膜在溫度較低時不易被溶解分散,影響維生素溶出效率,所以根據實驗結果選擇最佳提取溫度為65℃。
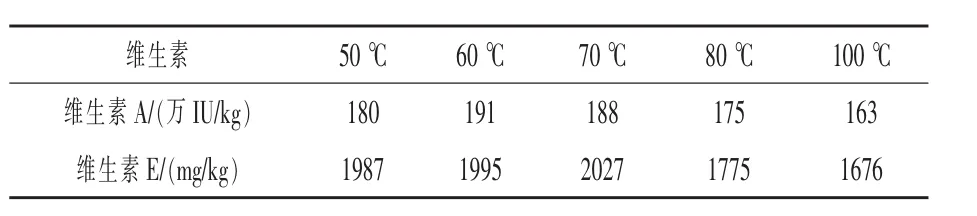
表4 皂化溫度對維生素測定結果的影響
2.6 皂化時間確定 取A,B兩批不同類型飼料樣品,每批7份,在65℃水浴中分別超聲5~120 min,其他條件按照1.3方法,計算含量,結果如表5,在120 min內提取的維生素含量基本不變,性質較穩定。考慮到其他飼料中維生素含有不同的包被,皂化時間太短不容易完全釋出,因此選擇20 min作為最終皂化時間。
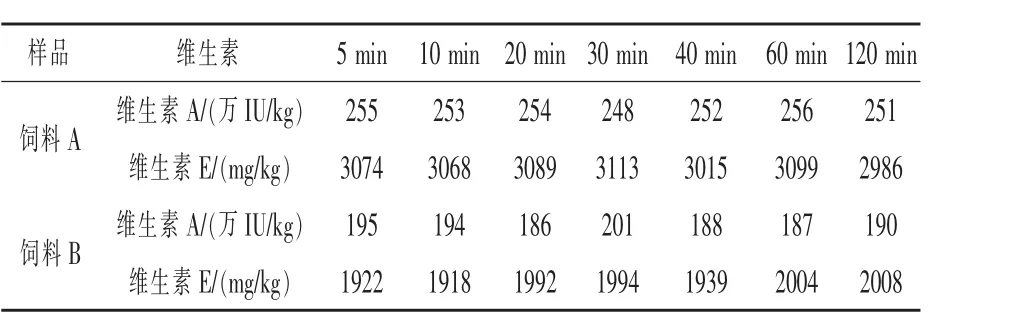
表5 皂化時間對維生素測定結果的影響
2.7 方法線性和檢測限 按配制好的系列標準工作液濃度從小到大,分別取20 μL注入液相色譜測定分析,記錄峰面積,以濃度(x)為橫坐標,峰面積(y)為縱坐標,繪制線性回歸曲線;同時添加適量的維生素A和維生素E標準儲備液至2 g空白飼料中,按照1.3方法處理,高效液相色譜分析并記錄色譜峰,以信噪比大于10確定定量限,結果見表6。

表6 維生素A和維生素E的標準曲線
2.8 回收率和精密度 取三批不同的空白飼料樣品,每批5份,每份2 g,分別添加適量的維生素A和維生素E標準儲備液,制得低、中、高三種濃度的樣品,按實驗方法1.3處理,取20 μL樣品溶液注入液相色譜測定分析,外標法計算含量,并計算回收率,結果見表7。
2.9 穩定性與重復性 取一份飼料樣品按照1.3方法處理,放置 0、1.5、3、4.5、10 h 后進樣測定,記錄維生素A和維生素E的峰面積并計算RSD分別為0.8%和1.1%,表明飼料樣品溶液中維生素A和維生素E在10 h內穩定。另取一批次飼料樣品6份,按照1.3方法處理,分別進樣測定,計算維生素A和維生素E含量,計算RSD分別為2.5%、4.9%,表明本方法重復性良好。
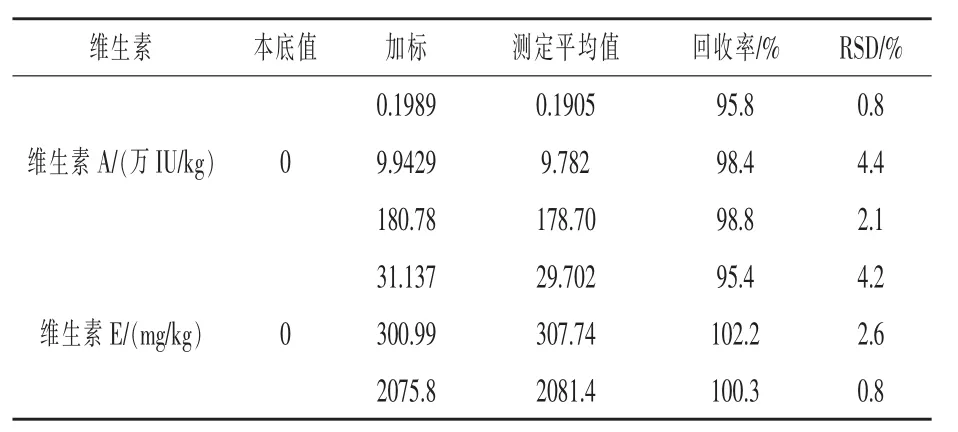
表7 回收率實驗結果(n=5)
2.10 與其他方法含量測定結果的比較 選取3種維生素預混料、5種復合預混料、1種配合料、1種濃縮料共10批次飼料樣品,分別用酶解法(曹冶,1997)、甲醇直接提取法(GB/T 17817-2010 第二法和GB/T 17812-2008第二法)、皂化法(GB/T 17817-2010第一法和GB/T 17812-2008第一法)和本方法處理測定維生素A和維生素E含量,結果如表8。酶解法測定時,不同樣品提取效果會有差異,一些樣品不能完全被提取,這可能和維生素包被壁材的類型有關系;甲醇提取法測定維生素A普遍偏低,測定維生素E相對較好,只有個別樣品偏低,一是因為在甲醇溶劑中,長時間高溫使得維生素A產生了異構體,另一方面也可能是和維生素包被壁材的類型有關,一些包膜不能被甲醇破壞溶解,維生素不能完全釋出;本方法和皂化法測定結果基本一致,略有偏高(特別是維生素E),這可能是本方法相對較少的步驟和時間,相對較低的皂化溫度,優化了皂化體系,減少了維生素的損失。因此在日常飼料檢測中可以利用本方法快速測定維生素A和E的含量,結果準確可靠。
3 討論與結論
目前維生素的壁材包被物質主要有明膠、阿拉伯膠、環糊精、變性淀粉、乳糖、玉米糖漿等以及多種壁材的混合物。淀粉和環糊精在高溫和堿的作用下易產生糊化作用分散在含水溶劑中形成膠體溶液;乳糖、玉米糖漿在水中易溶解;明膠、阿拉伯膠和乳清濃縮蛋白等含蛋白質物質在強堿加熱條件下容易水解。因此本方法采用皂化法,并且精簡優化操作,適用性廣,結果更準確,可以廣泛應用于飼料中維生素A和維生素E的質量控制。
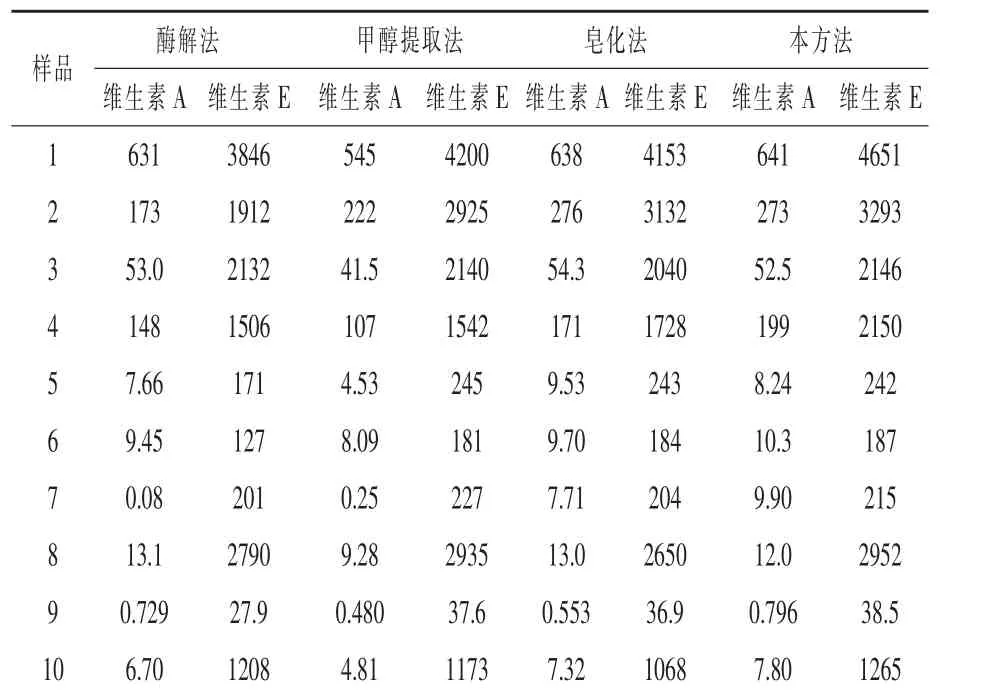
表8 四種方法測定飼料中維生素A和維生素E含量的比較
維生素預混料和復合預混料中主要成分是礦物質或者含纖維素多的物料,不含或含有極少量脂肪和蛋白質,皂化后的產物與其他有機質比較相對單一,采取皂化后直接進行色譜分析,皂化溫度相對較低,簡化了萃取操作,不僅提高工作效率,還減少樣品損失,降低了誤差。但是本方法取樣量相對國標皂化法少,對于飼料樣品的均勻度要求相對較高。
樣品提取液溶解有部分醋酸鉀等無機鹽和其他水溶物,在分析時也會進入到液相色譜系統。特別是其他水溶物,和流動相混合時,容易析出甲醇不溶物,因此在提取時減少鹽和水的含量,但又能滿足皂化要求,加入堿和抗壞血酸的量都選擇較低水平,提取液體系中水的含量控制在10%左右。同時在色譜分析一段時間或者完成全部分析后要用15%左右的甲醇水溶液沖洗色譜柱至少60 min,有利于延長色譜柱的使用壽命。
實際飼料樣品中,維生素A含量的范圍跨度比較大,使得制備的樣品上機液濃度范圍跨度也大,在使用相對應寬濃度范圍(最高濃度和最低濃度相差2500倍)標準校準曲線進行定量時,高濃度或者低濃度樣品的校準濃度和實際濃度有一定的差異,導致定量不準確,因此可以采用高、低兩個濃度范圍分段做標準校準曲線。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
